Chapter 12 Mapping
We proceeded to read mapping for every libraries on the built reference, already partly annotated:
- Repeats merging: 41 libraries were repeated, we merged their alignment to increase their information before variant calling
- Reads mapping: we mapped every libraries in pair end with
bwa memon the hybrid reference from Ivan Scotti et Sana Olson used to built the targets - Reference sequences: we built bedtools for every alignment in order to list sequences with mathces in the reference to be used to reduce explred reference area in variant calling
12.1 Repeats merging
41 libraries were repeated, we merged their FASTQ to increase their information before variant calling. Meging repeats confirmed the presence of all 430 individuals at the end of the alignment (402 from Paracou, 20 from herbariums, and 8 from BCI, Itubera and La Selva).
#!/bin/bash
#SBATCH --time=36:00:00
#SBATCH -J compression
#SBATCH -o compression_output.out
#SBATCH -e compression_error.out
#SBATCH --mem=4G
#SBATCH --cpus-per-task=1
#SBATCH --mail-type=BEGIN,END,FAIL
folder=trimmed.paired.joined/
name=symcapture.trimmed.paired.joined
module purge
mkdir trimmed.paired
for file in $(ls paired/*)
do
mv $file trimmed.paired/$(basename $(echo $file | sed -e 's/_[[:alpha:]]*-[[:alpha:]]*-[[:alnum:]]*//'))
done
rm -r paired
cp ../libraries.txt ./
cat libraries.txt | sed -e 's/_[[:alpha:]]*-[[:alpha:]]*-[[:alnum:]]*_L00[56]//' | sort | uniq | sed -e 's/-b//' | sort | uniq > libraries.uniq.txt
mkdir trimmed.paired.joined
for ind in $(cat libraries.uniq.txt)
do
echo $ind
cat trimmed.paired/$ind*_R1_paired.fq.gz > trimmed.paired.joined/"$ind"_R1_paired.fq.gz
cat trimmed.paired/$ind*_R2_paired.fq.gz > trimmed.paired.joined/"$ind"_R2_paired.fq.gz
done
rm -r trimmed.paired
tar -zcvf $name.tar.gz $folder12.2 Reads mapping
We mapped every libraries in pair end with bwa mem on the hybrid reference from Ivan Scotti et Sana Olson used to built the targets (parralelizing with 32 alignement with 2 process on 64 cores of 1 node of genologin). We had globally a good mapping with more than 80% of the reads mapped for 98% of the libraries 12.1.
#!/bin/bash
#SBATCH --time=48:00:00
#SBATCH -J mapping
#SBATCH -o mapping_output.out
#SBATCH -e mapping_error.out
#SBATCH --mem=160G
#SBATCH --cpus-per-task=64
#SBATCH --mail-type=BEGIN,END,FAIL
module purge
module load bioinfo/bwa-0.7.15
module load bioinfo/picard-2.14.1
module load bioinfo/samtools-1.4
module load bioinfo/bedtools-2.26.0
task(){
echo MAPPING "$1"
bwa mem -M -R "@RG\tID:$1\tSM:$1\tPL:HiSeq4K" \
-t 2 \
reference/reference.fasta \
trimming/trimmed.paired.joined/"$1"_R1_paired.fq.gz \
trimming/trimmed.paired.joined/"$1"_R2_paired.fq.gz \
> mapping/sam/"$1".sam
rm trimming/trimmed.paired.joined/"$1"_R1_paired.fq.gz
rm trimming/trimmed.paired.joined/"$1"_R2_paired.fq.gz
java -Xmx4g -jar $PICARD SortSam \
I=mapping/sam/"$1".sam \
O=mapping/bam2/"$1".bam
SORT_ORDER=coordinate
rm mapping/sam/"$1".sam
samtools index mapping/bam2/"$1".bam
}
mkdir mapping/sam
mkdir mapping/bam2
N=32
(
for library in $(cat unmapped.txt)
do
((i=i%N)); ((i++==0)) && wait
task "$library" &
done
)
rm -r mapping/sam# folders
mkdir mappingStat
touch readsMappingStat.txt
# test
file=$(ls bam*/*.bam | head -n 1)
module load bioinfo/samtools-1.4 ; samtools flagstat $file | echo $file" "$(grep "mapped (") >> readsMappingStat.txt
cat readsMappingStat.txt
rm readsMappingStat.txt
touch readsMappingStat.txt
# sarray
for file in $(ls bam*/*.bam); do echo 'module load bioinfo/samtools-1.4 ; samtools flagstat '$file' | echo '$file'" "$(grep "mapped (") >> readsMappingStat.txt'; done > mappingStat.sh
sarray -J mappingStat -o mappingStat/%j.out -e mappingStat/%j.err -t 1:00:00 --mail-type=BEGIN,END,FAIL mappingStat.sh
# clean
rm -r mappingStat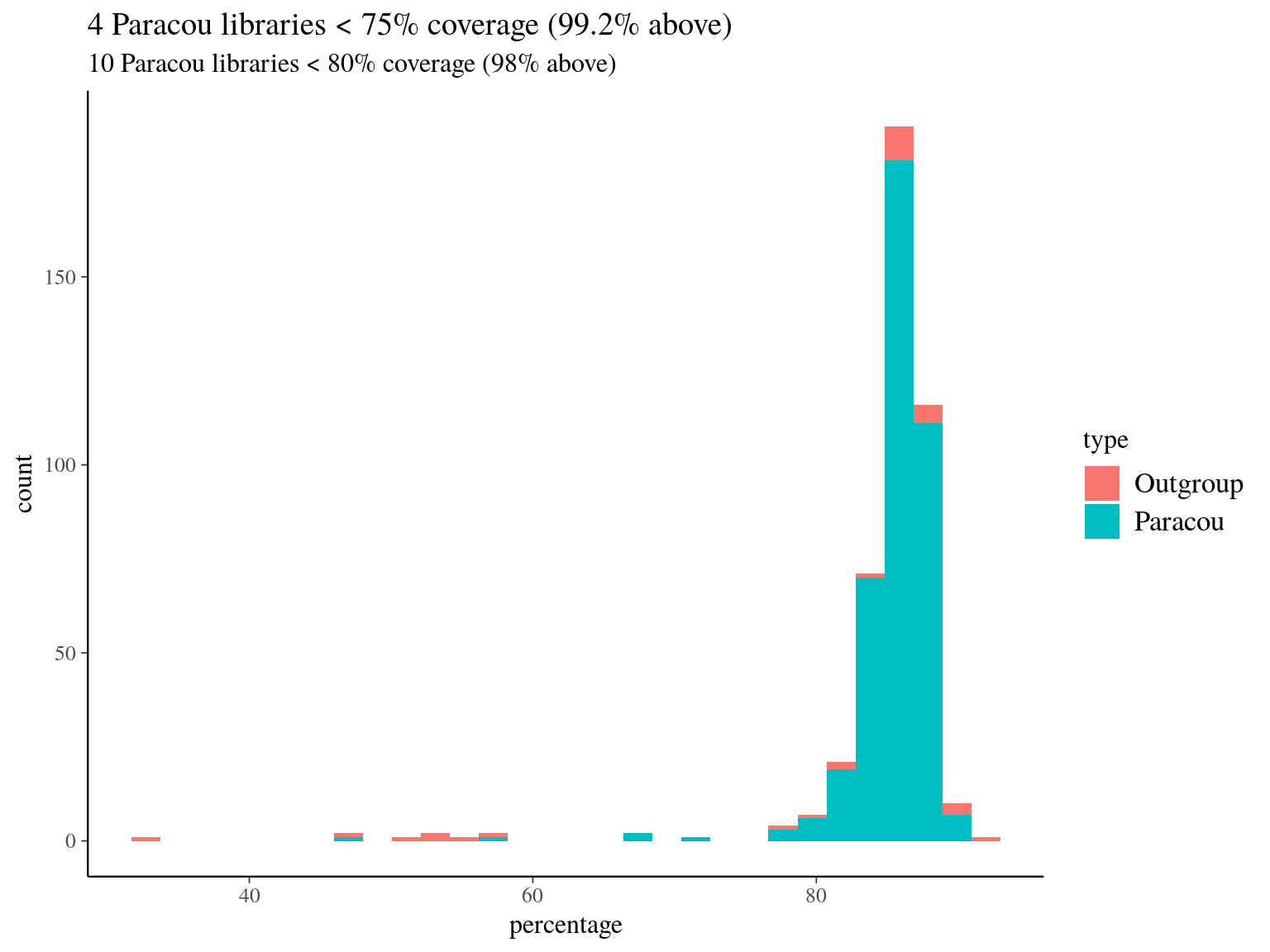
Figure 12.1: Mapping result
12.3 Reference sequences
We built bedtools for every alignment in order to list sequences with matches in the reference to be used to reduce explred reference area in variant calling. 99.98% of reference sequences have at least one library matching (12.2). Consequently we will use all sequences from the reference in the variant calling, besides few sequences our libraries are under represented, but they will be removed at the SNP filtering.
# folders
mkdir bed
mkdir bed.out
# test
file=$(ls bam*/*.bam | head -n 1)
module load bioinfo/bedtools-2.26.0 ; bedtools bamtobed -i $file > bed/$(basename "${file%.*}").bed
rm bed/*
# sarray
for file in $(ls bam*/*.bam); do echo 'module load bioinfo/bedtools-2.26.0 ; file='$file' ; bedtools bamtobed -i $file > bed/$(basename "${file%.*}").bed'; done > bed.sh
sarray -J bed -o bed.out/%j.out -e bed.out/%j.err -t 1:00:00 --mail-type=BEGIN,END,FAIL bed.sh
# clean
rm -r bed.out
# statistics
mkdir bed.out
touch referenceMappedStats.txt
for file in $(ls bed/*.bed); do echo "cut $file -f1 | sort | uniq | awk -v file="$(basename "${file%.*}")" '{print \$1, file}' >> referenceMappedStats.txt" ; done > bed.sh
sarray -J bed -o bed.out/%j.out -e bed.out/%j.err -t 1:00:00 --mail-type=BEGIN,END,FAIL bed.sh
rm -r bed.out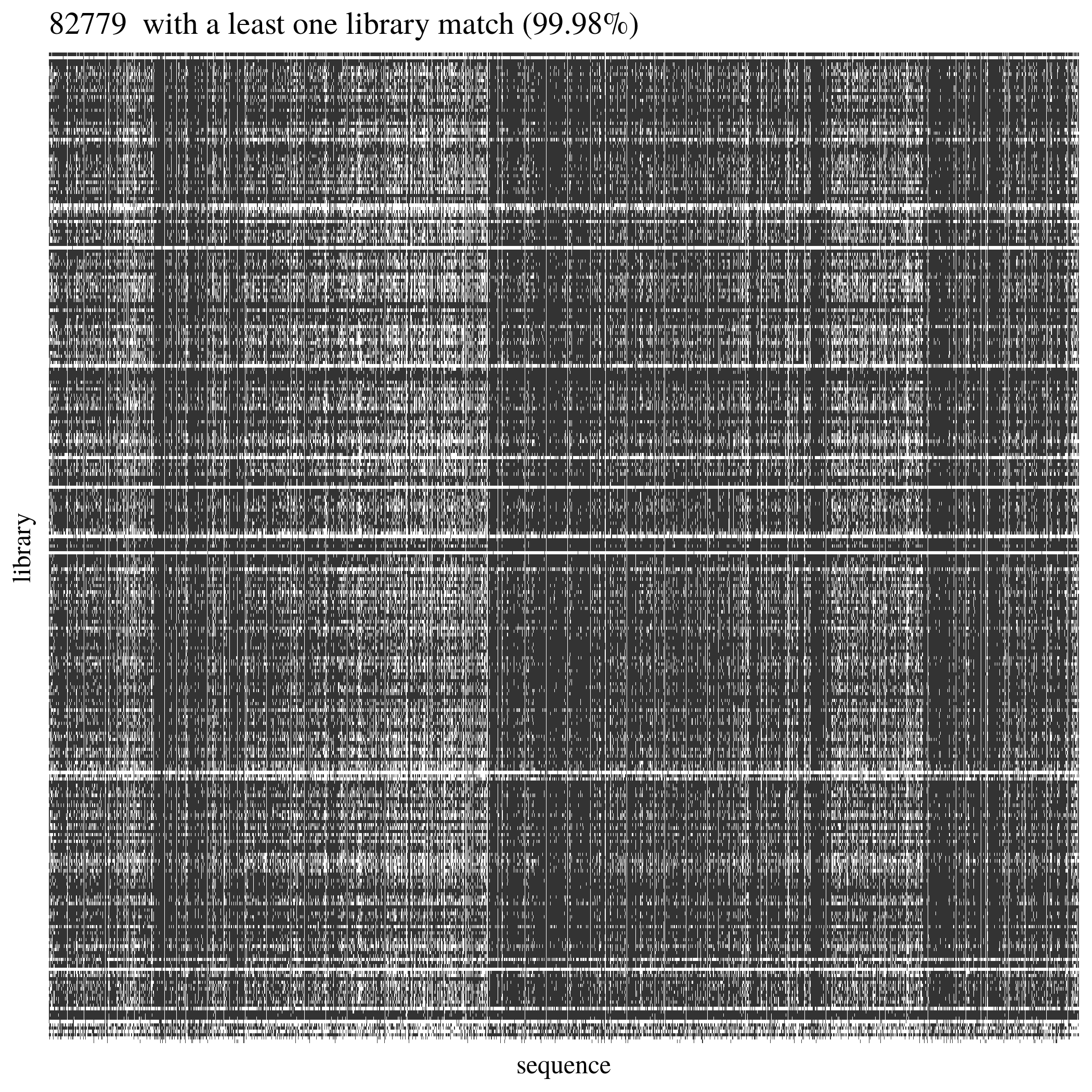
Figure 12.2: Sequences from reference alignment with reads from library.