Chapter 14 Variant filtering
We filtered the previously produced raw vcf with several steps:
- Gather raw vcf files gathering in 26 813 513 variants over 432 individuals
- Biallelic raw vcf filtering in 19 242 294 variants over 432 individuals
- SNP biallelic vcf filtering in 17 521 879 variants over 432 individuals
- Filters biallelic snp vcf filtering in 15 531 866 variants over 432 individuals
- Missing filtered biallelic snp vcf filtering in 454 262 variants over 406 individuals
- Paracou filtered & non missing biallelic snp vcf filtering in 454 262 variants over 385 individuals
14.1 Gather
We first gathered all raw vcf. 42 sequences subset lost individuals in genotype call (over 878, 4%), blocking the functionning of gatk CombineVariants, so we first removed the variants associated to those subest of reference sequences. This resulted in 26 813 513 variants.
mkdir out
mkdir missing_ind
for file in $(ls symcapture.filtered.vcf/*.vcf.gz) ; do file=$(basename $file) ; file=${file%.*} ; echo "module load bioinfo/tabix-0.2.5 ; module load bioinfo/vcftools-0.1.15 ; vcftools --gzvcf symcapture.filtered.vcf/$file.gz --missing-indv -c > missing_ind/$file.missing.txt"; done > missingInd.sh
sarray -J missingInd -o out/%j.missingInd.out -e out/%j.missingInd.err -t 1:00:00 --mail-type=BEGIN,END,FAIL missingInd.sh
for file in $(ls missing_ind/*.missing.txt) ; do awk '{{if (NR!=1) print FILENAME"\t"$0}}' $file ; done > missingInd.txt
rm - r out
rm -r missing_indecho -e '#!/bin/bash\n#SBATCH --time=48:00:00\n#SBATCH -J gather\n#SBATCH -o gather.out\n#SBATCH -e gather.err\n#SBATCH --mem=20G\n#SBATCH --cpus-per-task=1\n#SBATCH --mail-type=BEGIN,END,FAIL\nmodule load bioinfo/picard-2.14.1\njava -Xmx20g -jar $PICARD GatherVcfs \' > gather.sh
for file in $(cat nonmissing.list)
do
echo -e '\tI=symcapture.raw.vcf/'$(basename $file)' \' >> gather.sh
done
echo -e '\tO=symcapture.all.raw.vcf.gz\n' >> gather.sh
zcat symcapture.all.raw.vcf.gz | grep "#contig" | wc -l14.2 Biallelic
We then used bcftools to limit data to biallelic variants (--max-alleles 2), resulting in 19 242 294 biallelic variants.
14.3 SNP
We then used gatk to limit data to biallelic snps, resulting in 17 521 879 biallelic snps.
module load bioinfo/gatk-4.1.2.0
gatk IndexFeatureFile \
-F symcapture.all.biallelic.vcf.gz
gatk SelectVariants \
-V symcapture.all.biallelic.vcf.gz \
-select-type SNP \
-O symcapture.all.biallelic.snp.vcf.gz
gatk IndexFeatureFile \
-F symcapture.all.biallelic.snp.vcf.gz
module load bioinfo/bcftools-1.8
bcftools stats --threads 8 symcapture.all.biallelic.snp.vcf.gz14.4 Filters
We filtered the biallelic snp vcf with following filters (name, filter, description), resulting in 15 531 866 filtered biallelic snps, using next histograms to set and test parameters values :
- Quality (QUAL)
QUAL < 30: represents the likelihood of the site to be homozygote across all samples, we filter out variants having a low quality score (14.1) - Quality depth (QD)
QD < 2: filter out variants with low variant confidence (14.1) - Fisher strand bias (FS)
FS > 60: filter out variants based on Phred-scaled p-value using Fisher’s exact test to detect strand bias (14.1) - Strand odd ratio (SOR)
SOR < 3: filter out variants based on Phred-scaled p-value used to detect strand bias (14.1)
vcftools --gzvcf reference.sequences_00.vcf.gz --missing-indv -c
vcftools --gzvcf reference.sequences_00.vcf.gz --missing-site -c > missing.txt
vcftools --gzvcf reference.sequences_00.vcf.gz --site-quality -c > QUAL.txt
vcftools --gzvcf reference.sequences_00.vcf.gz \
--get-INFO AC \
--get-INFO AF \
--get-INFO QD \
--get-INFO FS \
--get-INFO SOR \
-c > INFO.txt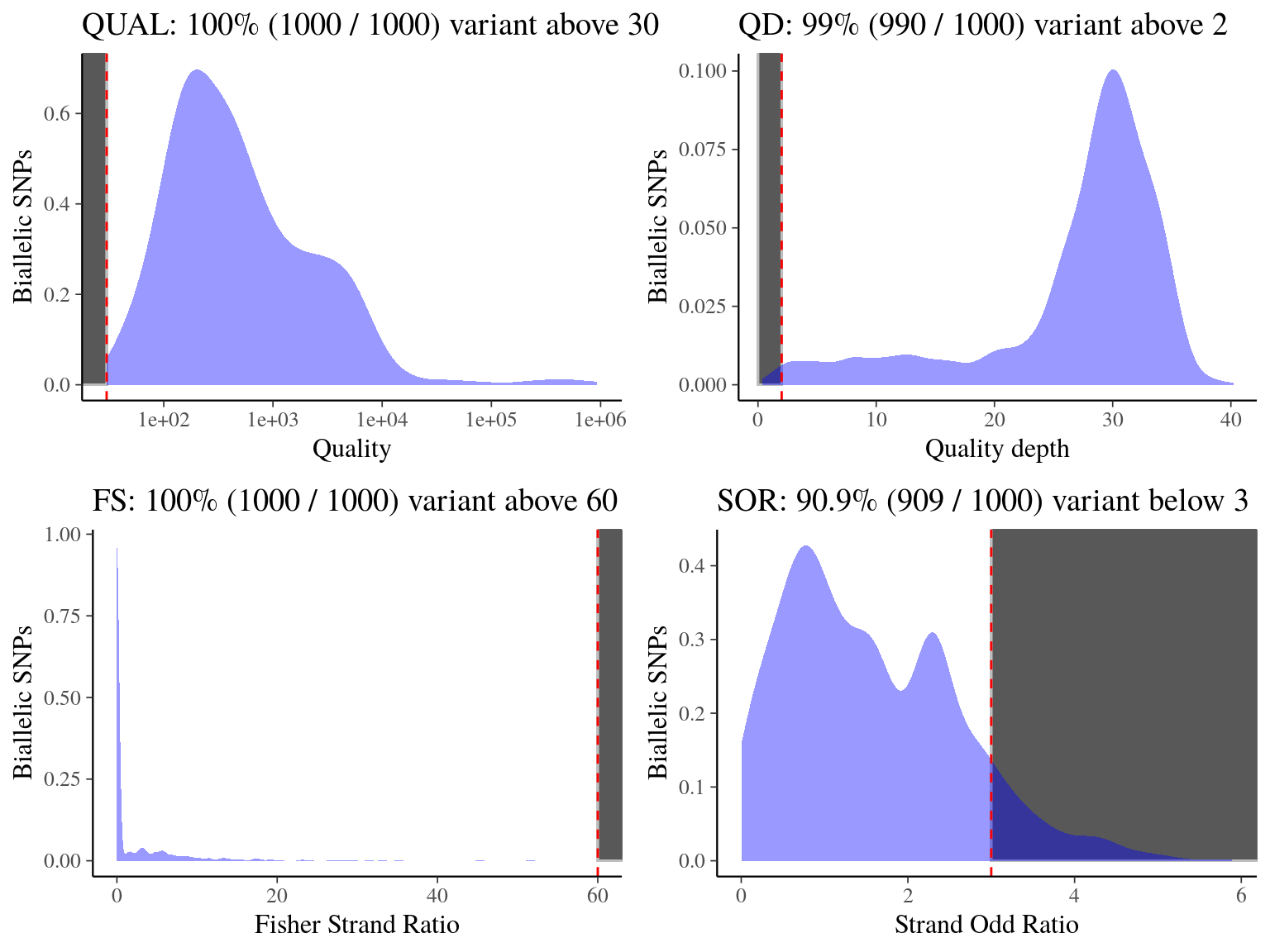
Figure 14.1: Quality, quality by depth, fisher strand and strand odd ratios per biallelic SNPs.
module load bioinfo/gatk-4.1.2.0
gatk VariantFiltration \
-V symcapture.all.biallelic.snp.vcf.gz \
--filter-expression "QUAL < 30.0 || QD < 2.0 || FS > 60.0 || SOR > 3.0" \
--filter-name "FAIL" \
-O symcapture.all.biallelic.snp.intermediate.vcf.gz
gatk SelectVariants \
-V symcapture.all.biallelic.snp.intermediate.vcf.gz \
--exclude-filtered \
-O symcapture.all.biallelic.snp.filtered.vcf.gz
module load bioinfo/bcftools-1.8
bcftools stats --threads 8 symcapture.all.biallelic.snp.filtered.vcf.gz
gatk IndexFeatureFile \
-F symcapture.all.biallelic.snp.filtered.vcf.gz14.5 Missing data
Missing data filtering is a bit more tricky because missing data of SNPs and individuals are related, e.g. removing individuals with a lot of missing data result in the decrease of SNPs. Ideally, we would want to keep all individuals, but this would result in a lot of SNP loss because of least represented individuals. So we need to chose a threshold for missing data for individuals --mind and SNPs --geno.
module load bioinfo/plink_high_contig_20190905
module load bioinfo/plink2_high_contig_20190905
mkdir filtered
plink2 --threads 8 --memory 80000 \
--vcf symcapture.all.biallelic.snp.filtered.vcf.gz \
--allow-extra-chr \
--make-bed --out filtered/symcapture.all.biallelic.snp.filtered
cd filtered
plink --threads 8 --memory 80000 \
--bfile symcapture.all.biallelic.snp.filtered \
--allow-extra-chr --missing --het --freq --pca --freqx \
--out symcapture.all.biallelic.snp.filtered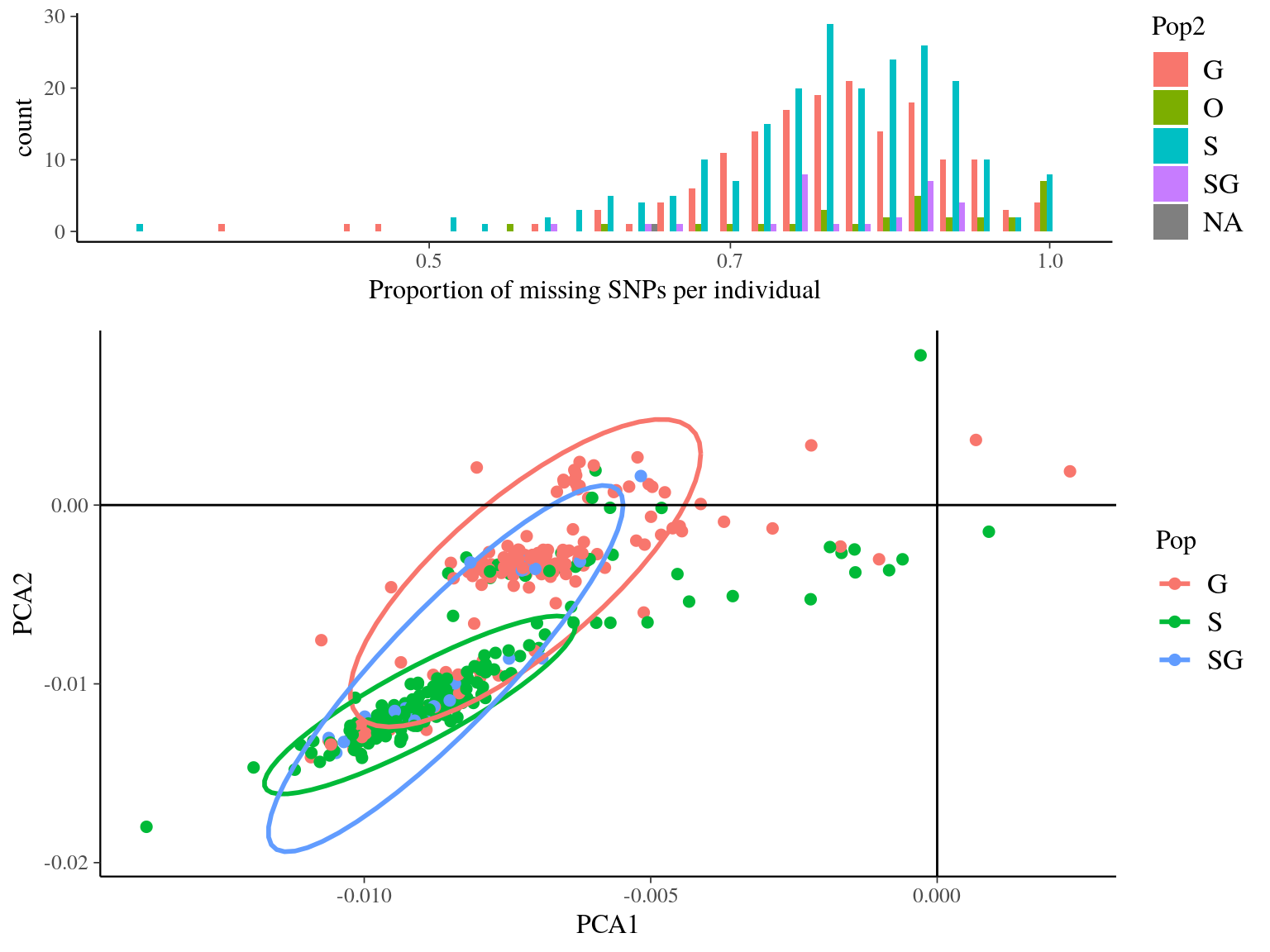
Figure 14.2: Missing data statistics for filtered biallelic SNP before missing data filtering per individual.
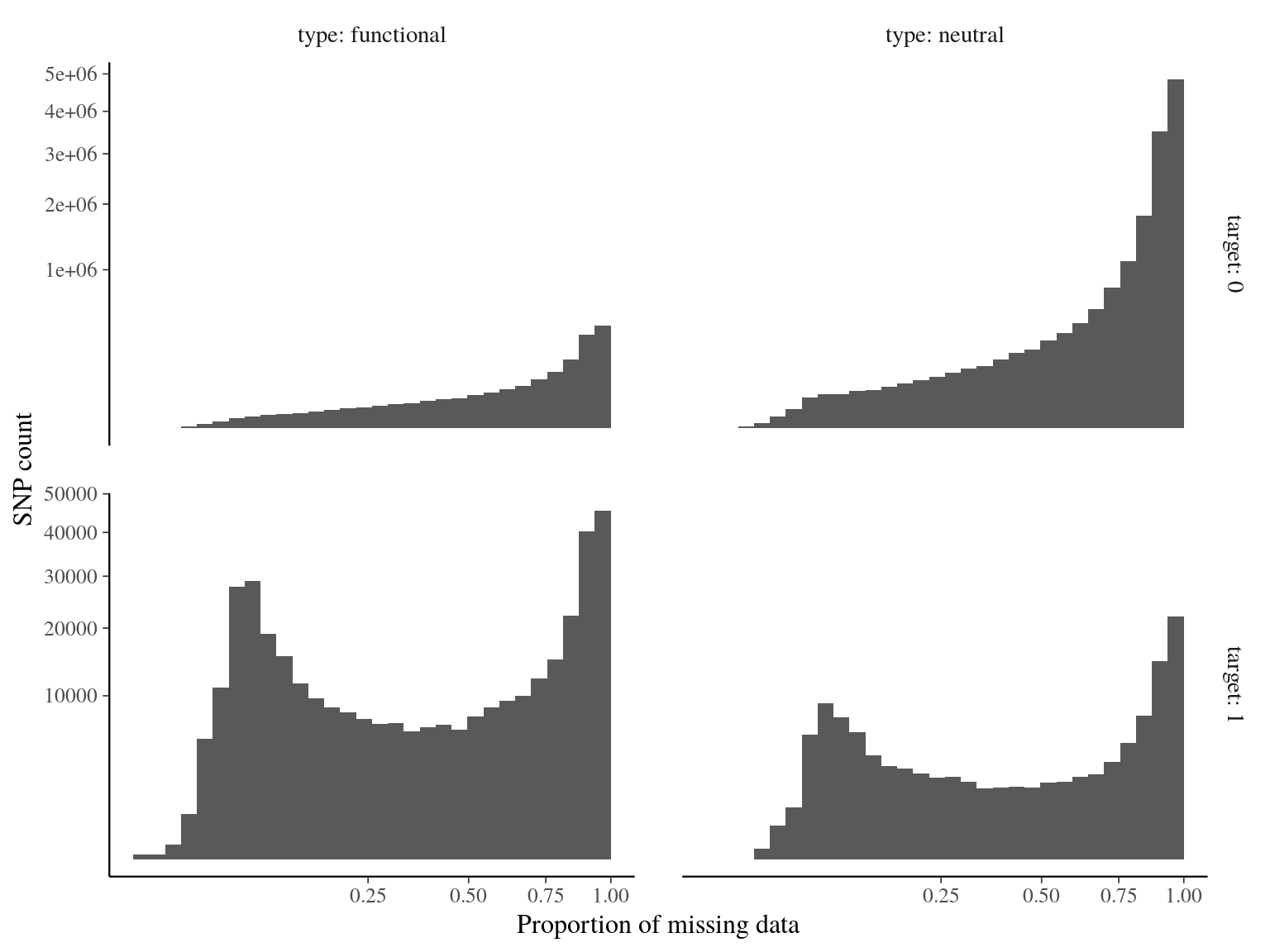
Figure 14.3: Missing data statistics for filtered biallelic SNP before missing data filtering per SNP.
14.5.1 Normal filter
With a maximum of 95% of missing data per individual --mind 0.95 and a maximum of 15% of missing data per SNP -geno 0.15, we obtained 454 262 biallelic filtered snps for 406 individuals.
module load bioinfo/plink_high_contig_20190905
module load bioinfo/plink2_high_contig_20190905
mkdir nonmissing
plink2 --threads 8 --memory 80000 \
--bfile filtered/symcapture.all.biallelic.snp.filtered \
--allow-extra-chr \
--mind 0.95 --geno 0.15 \
--make-bed --out nonmissing/symcapture.all.biallelic.snp.filtered.nonmissing
cd nonmissing
plink --threads 8 --memory 80000 \
--bfile symcapture.all.biallelic.snp.filtered.nonmissing \
--allow-extra-chr --missing --het --freqx --pca \
--out symcapture.all.biallelic.snp.filtered.nonmissing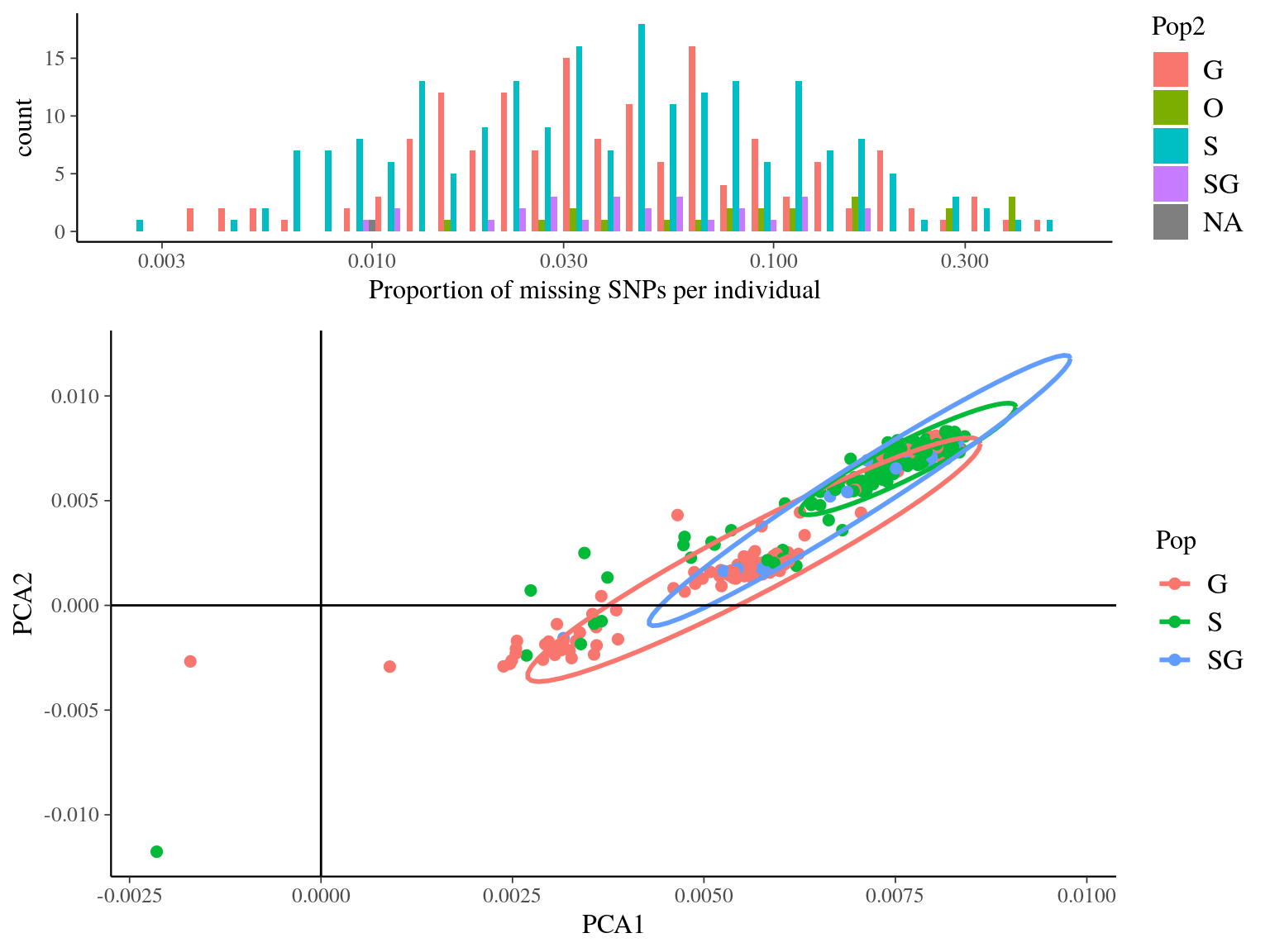
Figure 14.4: Missing data statistics for filtered biallelic SNP after missing data filtering (95% for individuals and 15% for SNPs) per individual.
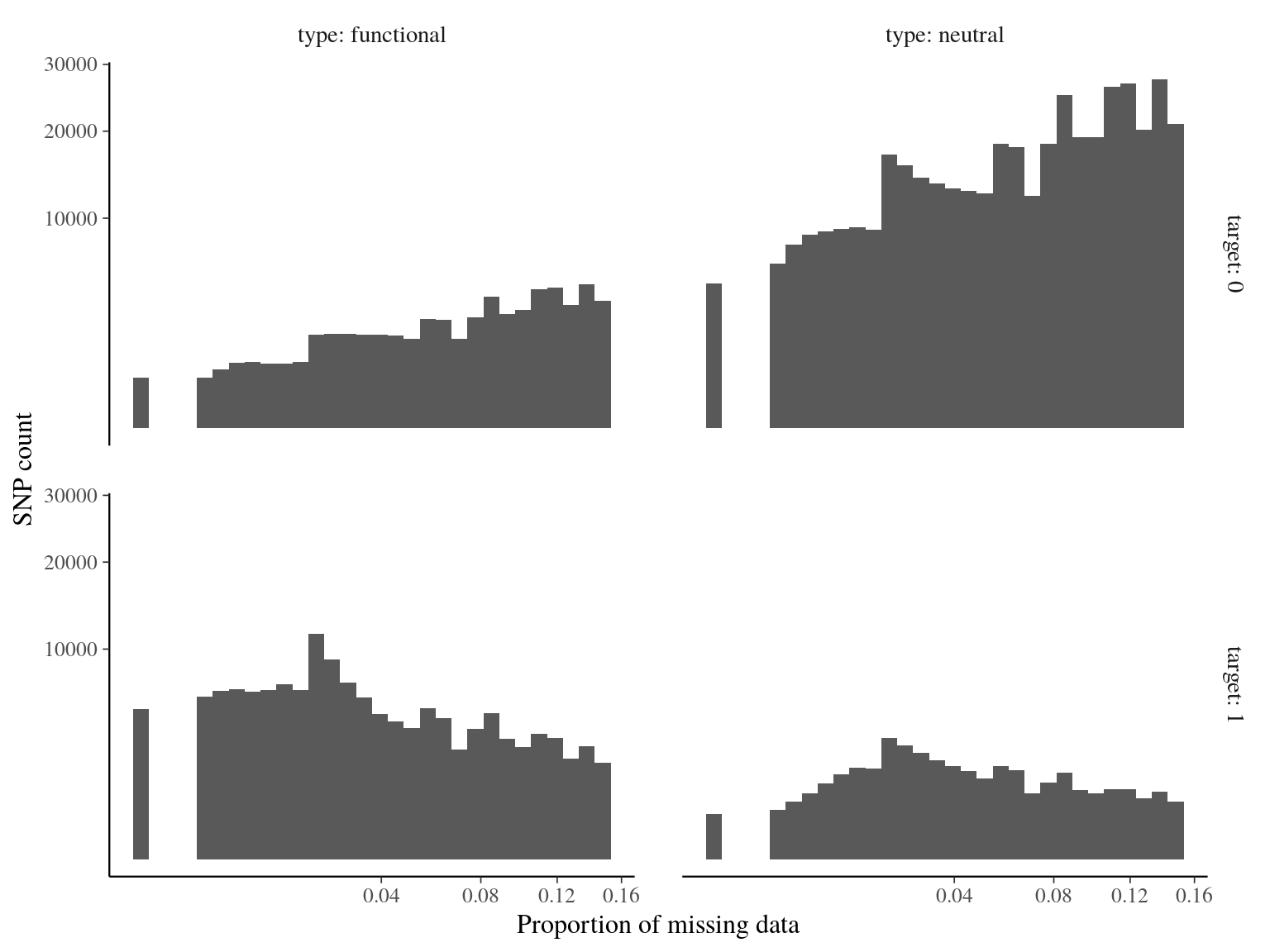
Figure 14.5: Missing data statistics for filtered biallelic SNP after missing data filtering (95% for individuals and 15% for SNPs) per SNP.
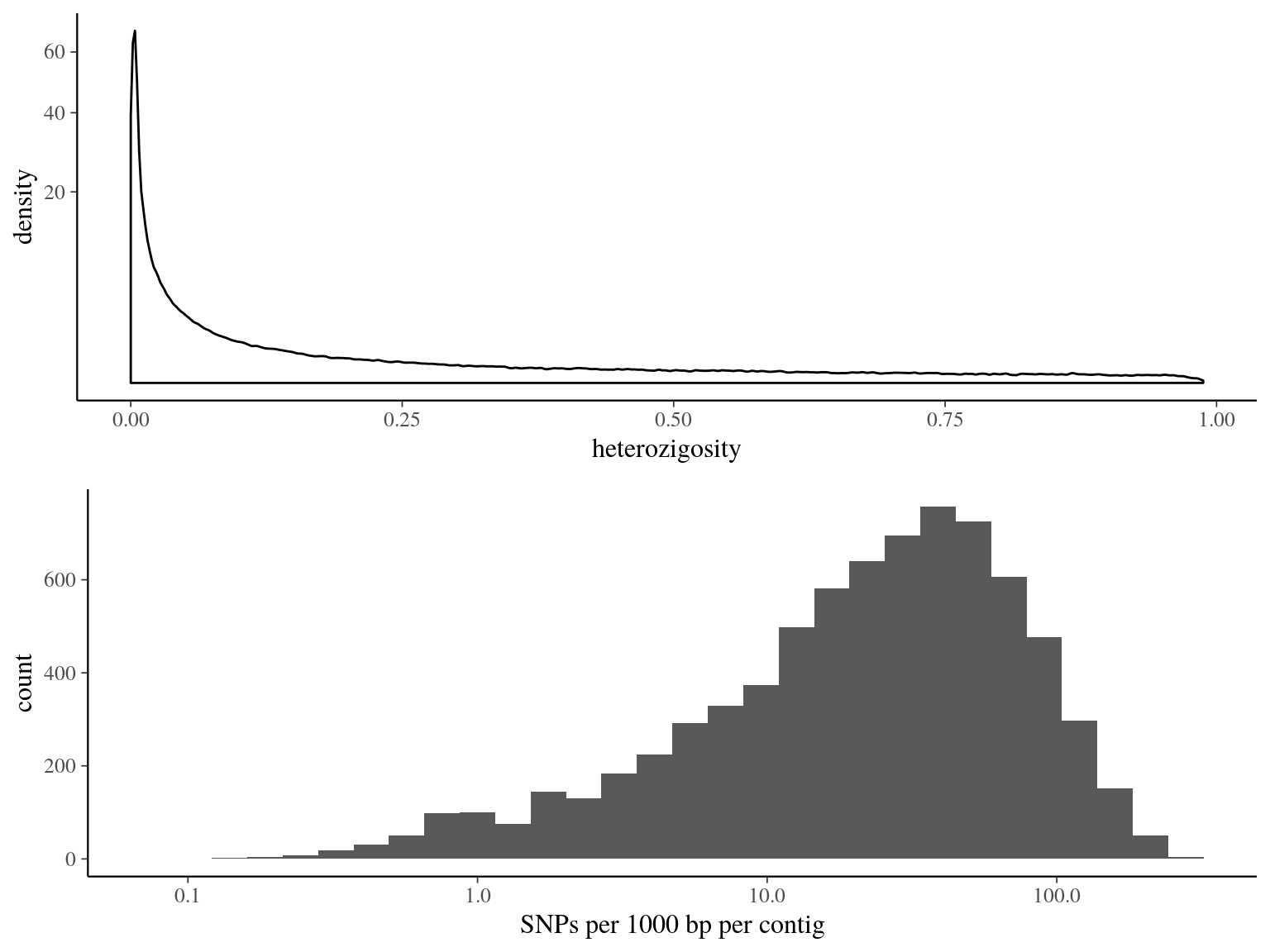
Figure 14.6: Heterozigosity statistics for filtered biallelic SNP after missing data filtering (95% for individuals and 15% for SNPs) per SNP.
14.5.2 Hard filter
With a maximum of 95% of missing data per individual --mind 0.95 and a maximum of 5% of missing data per SNP -geno 0.05, we obtained 180 217 biallelic filtered snps for 406 individuals.
module load bioinfo/plink_high_contig_20190905
module load bioinfo/plink2_high_contig_20190905
mkdir hardFilter
plink2 --threads 8 --memory 80000 \
--bfile filtered/symcapture.all.biallelic.snp.filtered \
--allow-extra-chr \
--mind 0.95 --geno 0.05 \
--make-bed --out hardFilter/symcapture.all.biallelic.snp.filtered.hardfiltered
cd hardFilter
plink --threads 8 --memory 80000 \
--bfile symcapture.all.biallelic.snp.filtered.hardfiltered \
--allow-extra-chr --missing --het --freq --pca \
--out symcapture.all.biallelic.snp.filtered.hardfiltered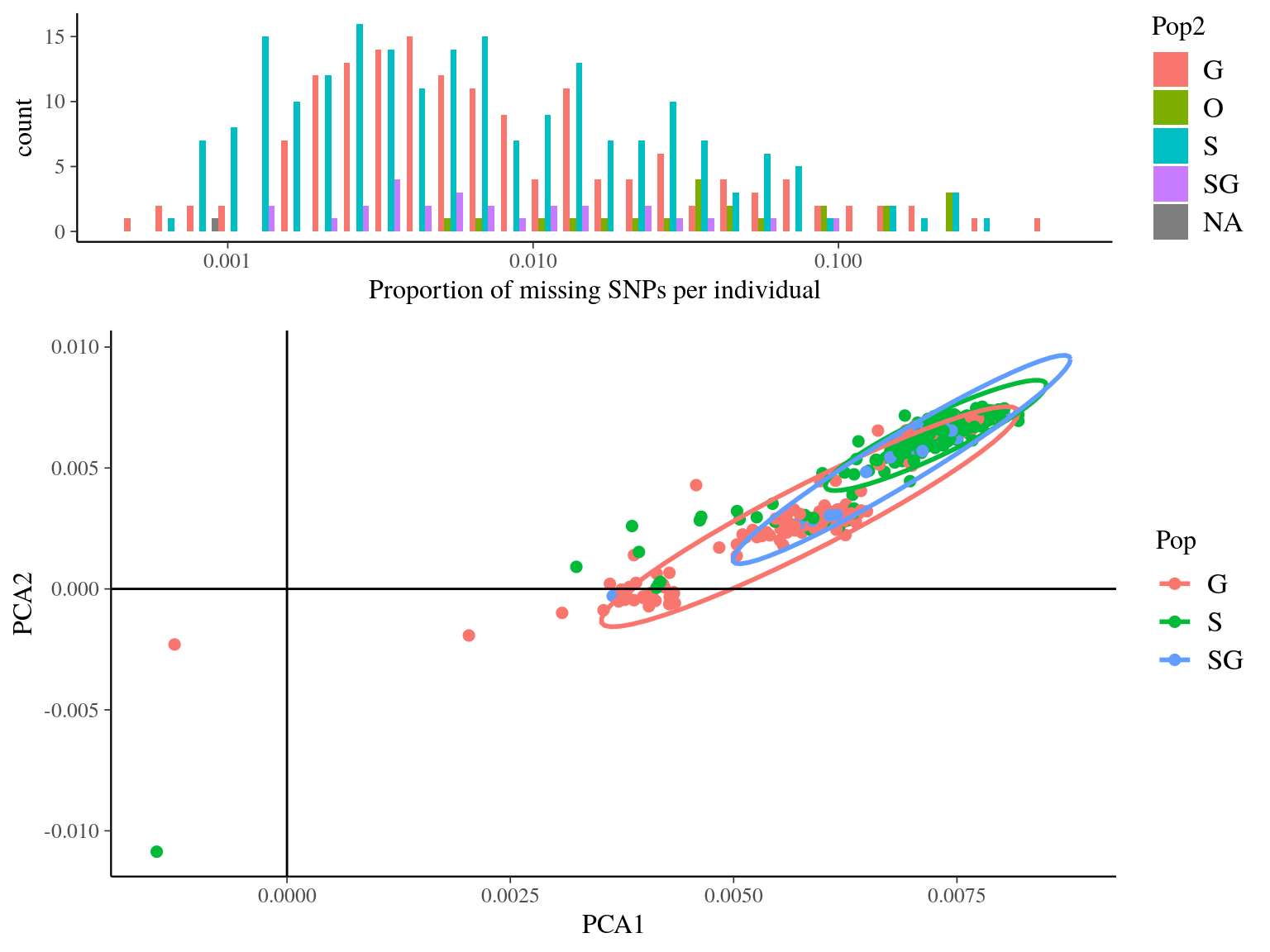
Figure 14.7: Missing data statistics for filtered biallelic SNP after missing data filtering (95% for individuals and 5% for SNPs) per individual.
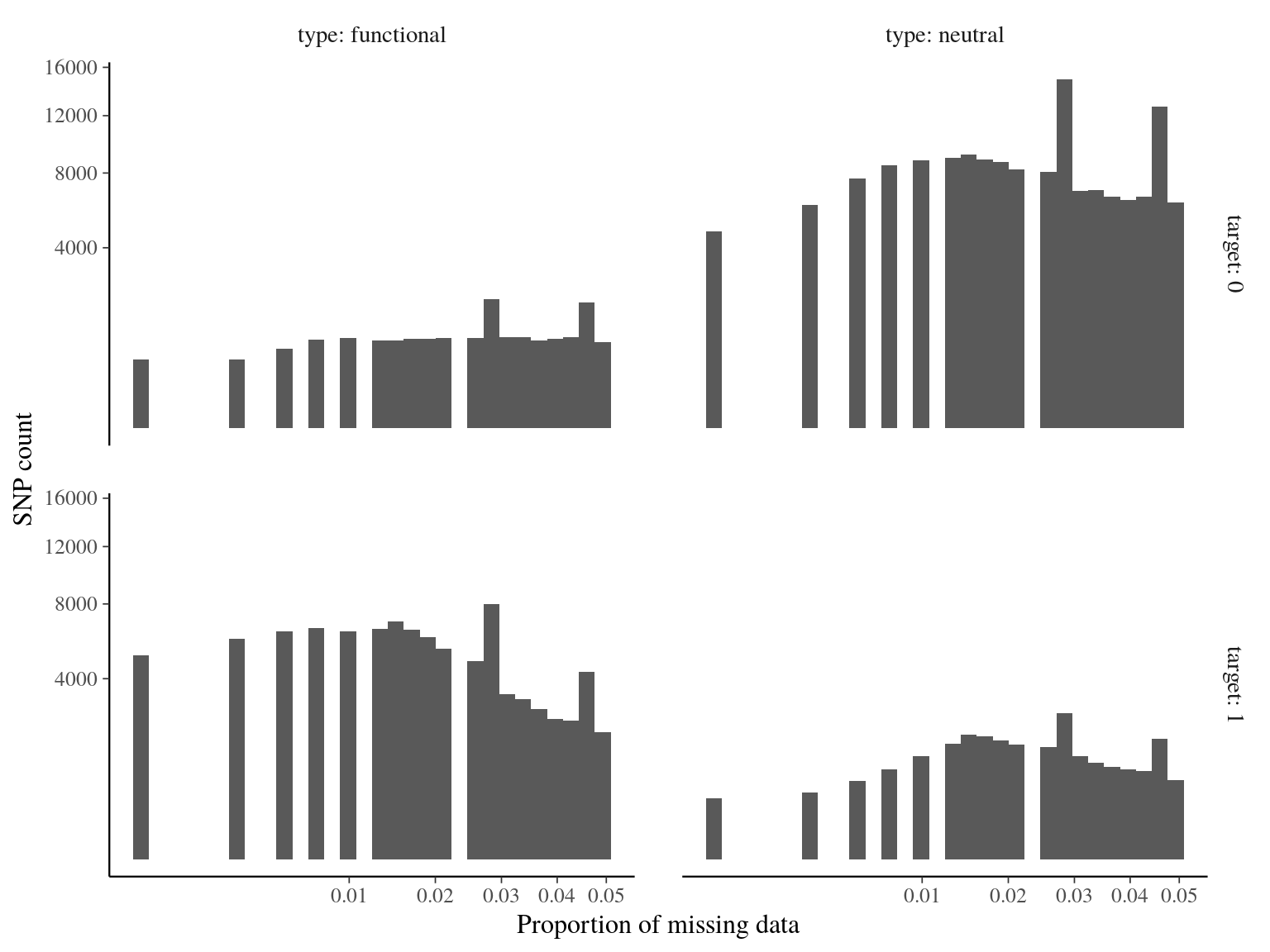
Figure 14.8: Missing data statistics for filtered biallelic SNP after missing data filtering (90% for individuals and 15% for SNPs) per SNP.
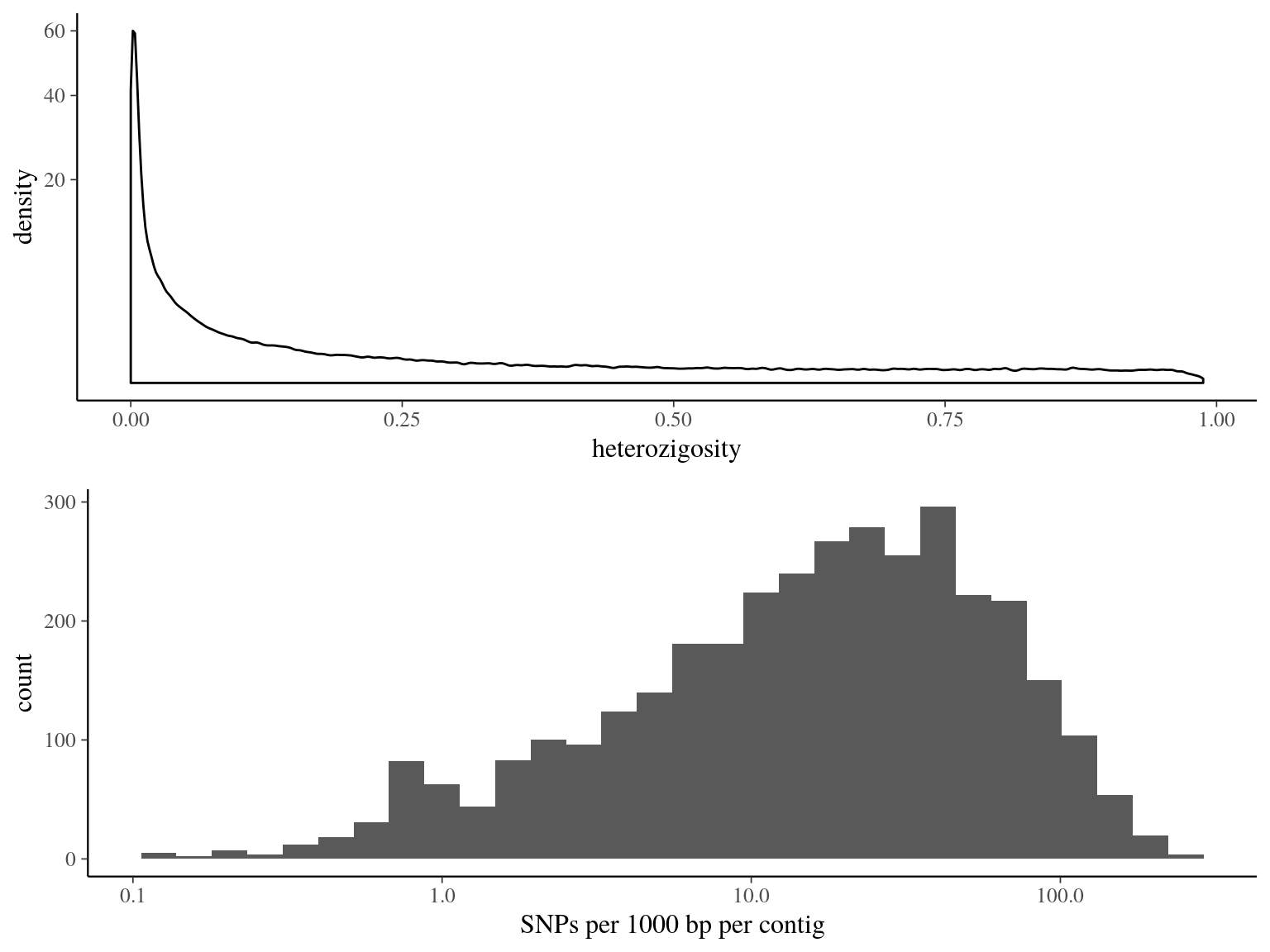
Figure 14.9: Heterozigosity statistics for filtered biallelic SNP after missing data filtering (95% for individuals and 5% for SNPs) per SNP.
14.6 Paracou
Finally, we subseted the filtered and biallelic snp from Paracou individuals, resulting in 385 remaining individuals (17 lost !).