Chapter 1 Scotti et al. (in prep) scaffolds preparation
The Guianan scaffolds from Scotti et al (in prep)…
1.1 Filtering scaffolds over \(1kbp\)
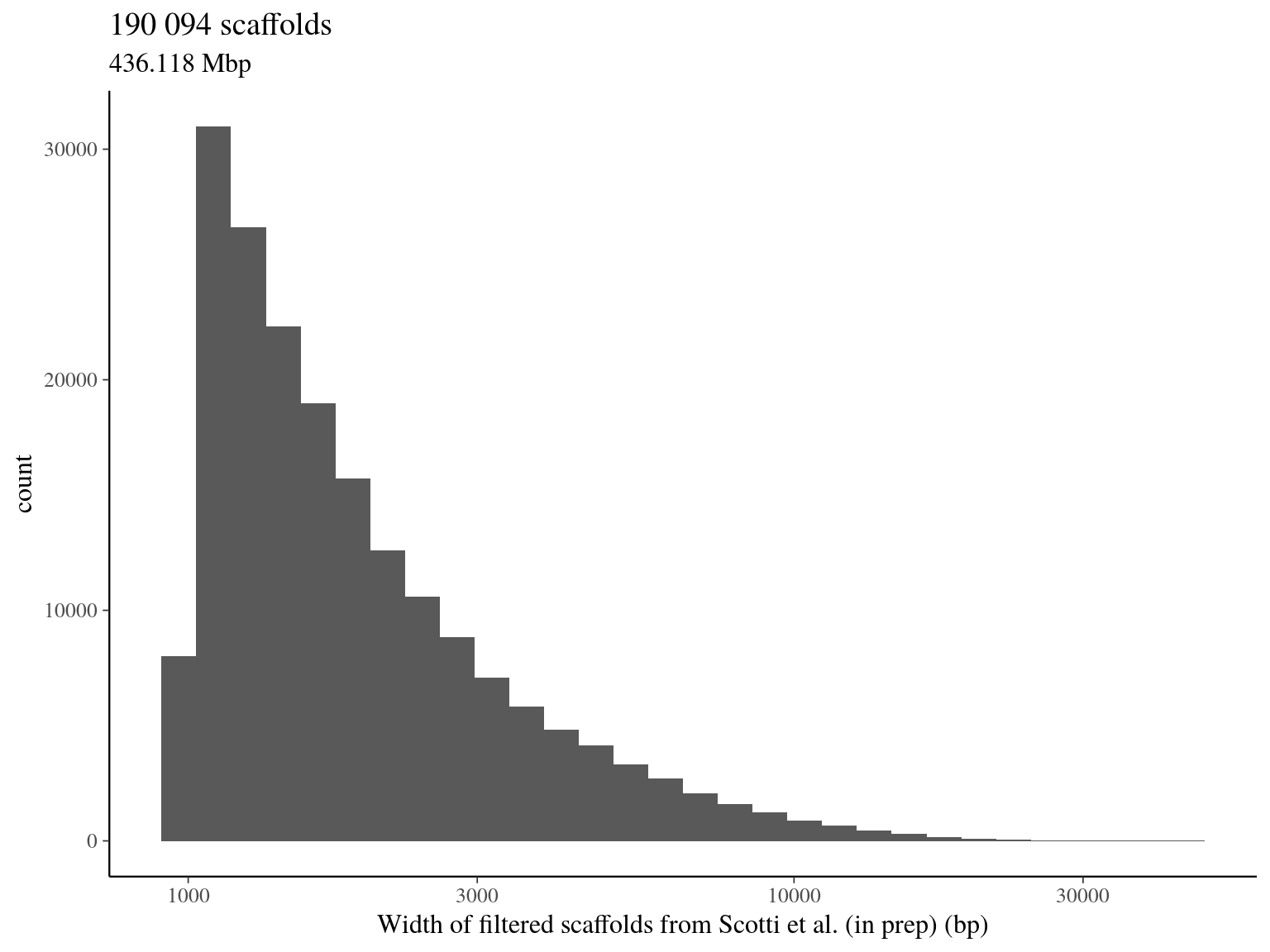
Because of the maybe poor quality of the assembly for scaffolds from Scotti et al (in prep), we first start by filtering scaffolds with a width superior to 1000 bp.
1.2 Renaming
For all scaffolds we will use following code : **Ivan_2018_[file name without .scafSeq]_[scaffold name]**.
dir.create(file.path(path, "Ivan_2018", "renamed_scf_1000"))
files <- list.files(file.path(path, "Ivan_2018", "scf_1000"))
sapply(files, function(file){
scf <- readDNAStringSet(file.path(path, "Ivan_2018", "scf_1000", file))
names(scf) <- paste0("Ivan_2018_", gsub(".scafSeq", "", file), "_", gsub(" ", "_", names(scf)))
writeXStringSet(scf, file.path(path, "Ivan_2018", "renamed_scf_1000",
paste0(gsub(".scafSeq", "", file), ".1000.renamed.scafSeq")))
}, simplify = F)
unlink(file.path(path, "Ivan_2018", "scf_1000"), recursive = T)1.3 Libraries merging
We will successively merge scaffolds from all libraries with quickmerge. We have 9 libraries resulting in 8 successive merge. We were only able to merge the 6 first libraries due to memory limiitation on my local machine and the absence of quickmerge on genotoul/genologin. We will still use this merging as a new reference.
cd ~/Documents/BIOGECO/PhD/data/Symphonia_Genomes/Ivan_2018/renamed_scf_1000
ref=(sympho47_1L1_002.1000.renamed.scafSeq merge1.fasta merge2.fasta merge3.fasta merge4.fasta merge5.fasta merge6.fasta merge7.fasta)
query=(sympho47_1L2_001.1000.renamed.scafSeq sympho47_2L1_008.1000.renamed.scafSeq sympho47_2L1_009.1000.renamed.scafSeq sympho47_2L1_010.1000.renamed.scafSeq sympho47_2L1_011.1000.renamed.scafSeq sympho47_2L1_012.1000.renamed.scafSeq sympho47_3L2_013.1000.renamed.scafSeq sympho47_4L1_014.1000.renamed.scafSeq)
for i in {0..7}
do
mkdir merge$i
cp "${ref[$i]}" "${query[$i]}" ./merge$i/
cd merge$i
nucmer -l 100 -prefix out ${ref[$i]} ${query[$i]}
delta-filter -i 95 -r -q out.delta > out.rq.delta
~/Tools/quickmerge/quickmerge -d out.rq.delta -q ${query[$i]} -r ${ref[$i]} -hco 5.0 -c 1.5 -l n -ml m
cd ..
cp merge$i/merged.fasta ./merge$((i+1)).fasta
done1.4 Removing scaffolds with multimatch blasted consensus sequence from Torroba-Balmori et al. (unpublished)
We used the consensus sequence for French Guianan reads from Torroba-Balmori et al. (unpublished) previously assembled with ipyrad. As a first brutal approach we only keep the first sequence of the consensus loci file and transform it to fasta (see loci2fa.py script below). We then blast those consensus sequences on merged scaffolds from Scotti et al (in prep) with blastn in order to detect scaffolds with repetitive regions thanks to multimapped consensus sequences. Those sequences will be saved as a list to be removed in final selected scaffolds list.
infile = open("symphoGbS2.loci", "r")
outfile = open("symphoGbS2.firstline.fasta", "w")
loci = infile.read().split("|\n")[:-1]
for loc in loci:
reads = loc.split("\n")
name, seq = reads[0].split()
print >>outfile, ">"+name+"\n"+seq
outfile.close()cd ~/Documents/BIOGECO/PhD/data/Symphonia_Torroba/assembly/symphoGbS2_outfile
python loci2fa.py
cat symphoGbS2.firstline.fasta | tr - N >> symphoGbS2.firstline.fastacd ~/Documents/BIOGECO/PhD/data/Symphonia_Genomes/Ivan_2018
query=~/Documents/BIOGECO/PhD/data/Symphonia_Torroba/assembly/symphoGbS2_outfiles/symphoGbS2.firstline.fasta
blastn -db merged_1000/merged_1000.fa -query $query -out blast_consensus_torroba2.txt -evalue 1e-10 -best_hit_score_edge 0.05 -best_hit_overhang 0.25 -outfmt 6 -perc_identity 75 -max_target_seqs 10blast <- read_tsv(file.path(path, "Ivan_2018", "blast_consensus_torroba2.txt"), col_names = F)
names(blast) <- c("Read", "Scaffold", "Perc_Ident", "Alignment_length", "Mismatches",
"Gap_openings", "R_start", "R_end", "S_start", "S_end", "E", "Bits")
write_file(paste(unique(blast$Scaffold), collapse = "\n"),
file.path(path, "Ivan_2018", "selected_scaffolds_blast_consensus2.list"))In total 542 scaffolds from Scotti et al (in prep) match consensus sequences from Torroba-Balmori et al. (unpublished). But several scaffolds obtained multiple matches that we cannot use for probes. We will thus exclude the whole scaffold if the scaffold is shorter than 2000 bp or the scaffold region matching the raw read if the scaffold is longer than 2000 bp.
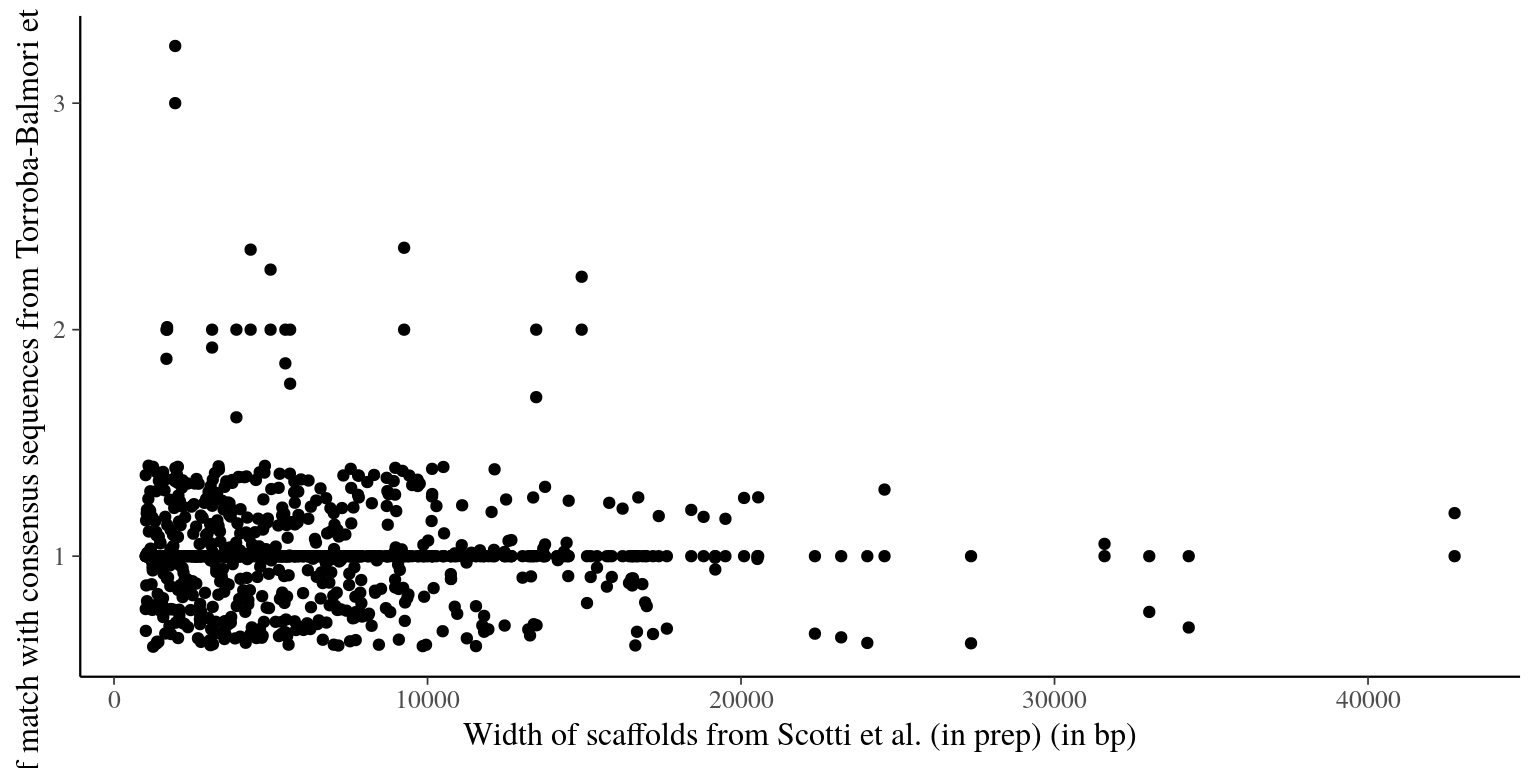
Figure 1.1: Number of match with Torroba consensus reads vs gene width.
| Scaffold | width | remove | cut |
|---|---|---|---|
| Ivan_2018_sympho47_2L1_012_scaffold197676__8.6 | 4993 | 4993-4932 | |
| Ivan_2018_sympho47_2L1_012_scaffold246452__6.6 | 3103 | 2980-3058 | |
| Ivan_2018_sympho47_2L1_012_scaffold26367__7.7 | 2168 | 2118-2168 | |
| Ivan_2018_sympho47_2L1_012_scaffold463128__4.7 | 2188 | 667-586 | |
| Ivan_2018_sympho47_2L1_008_scaffold309475__2.1 | 3525 | 342-292 |
Following scaffolds will be removed due to multitple matches and a length \(<200bp\): 2L1_012_scaffold645876__7.5, 2L1_012_scaffold176548__7.1, 2L1_012_scaffold21882__4.9, 2L1_012_scaffold9236__6.0. And other will be cut (see table 1.1).
1.5 Total filtered scaffolds