Chapter 3 Tysklind et al (in prep) transcript preparation
Tysklind et al (in prep) used 20 Symphonia juveniles from the transplantation garden experiment for transcriptomic analysis. RNA sequence were captured. The analysis followed the scheme suggested by Lopez-Maestre et al. (2016) (see below). First, reads were assembled with Trinity into transcripts. In parrallel, SNPs were detected with Kissplice. Then SNPs have been mapped on the transcritpome with BLAT. In parrallel SNPs have been tested to be morphotype-specific at the level \(\alpha = 0.001\) with KissDE and transcriptome Open Reading Frames (ORF) have been indentified with Transdecoder. Finally, SNPs functional impact have been evaluated through k2rt. Consequently, for every SNP we have the following informations: (i) inside coding DNA sequence (CDS), (ii) synonymous or not, (iii) morphotype-specificity.
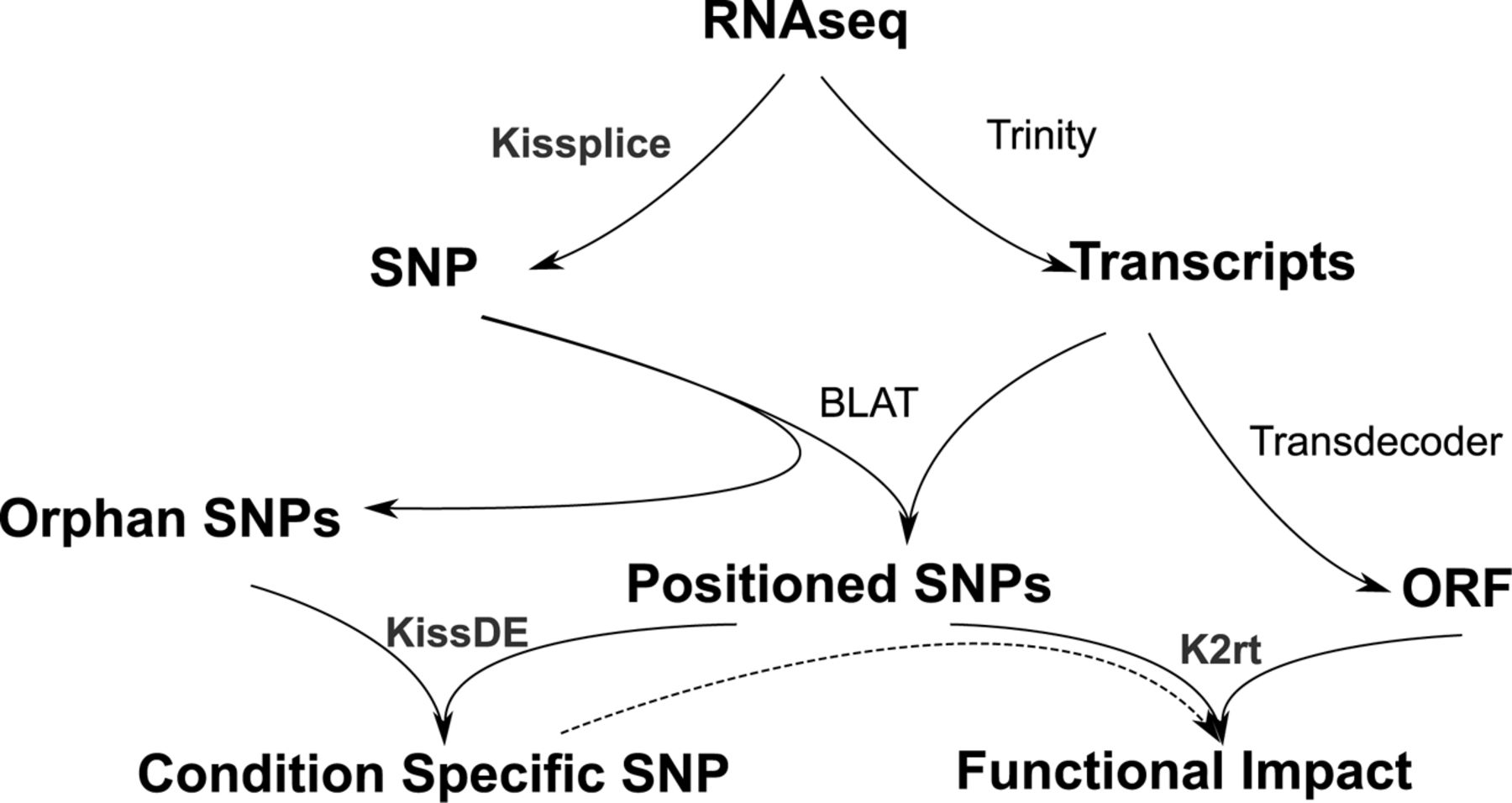
Analysis scheme from Lopez-Maestre et al. (2016).
3.1 Filtering SNP on quality
We assessed transcriptomic analysis quality with possible sequencing errors, and SNPs in multiple assembled genes or isoforms (see table 3.1). We found 38 594 SNPs with possible sequencing error, and 609 214 SNPs associated to multiple assembled genes that we will remove from further analysis.
| variable | n | Percentage |
|---|
3.2 Filtering SNP on type
We also highlighted SNPs which met unpossible association of characteristic (table 3.2), that we will remove from further analysis.
| Coding sequence | Not synonymous | Morphotype-specific | n | type |
|---|---|---|---|---|
| False | False | morphotype specific | 9 505 | unpossible |
| False | False | morphotype specific | 5 362 | unpossible |
| False | True | morphotype specific | 11 819 | unpossible |
| False | True | morphotype specific | 5 565 | unpossible |
| N/A | False | morphotype specific | 42 | unpossible |
| N/A | False | morphotype specific | 21 | unpossible |
| N/A | N/A | morphotype specific | 2 557 | unpossible |
| N/A | N/A | morphotype specific | 424 | unpossible |
| N/A | True | morphotype specific | 63 | unpossible |
| N/A | True | morphotype specific | 9 | unpossible |
| True | N/A | morphotype specific | 26 482 | unpossible |
| True | N/A | morphotype specific | 14 342 | unpossible |
3.3 Filtering transcripts on SNP frequency
We had a high frequency of SNPs per candidate genes (the majority between 1 SNP per 10 or 100 bp), with some scaffolds with a frquency superior to 0.2 (see figure 3.1). We assumed those hyper SNP-rich scaffolds to be errors and we decided to remove them of the reference transcriptome. In order to do that we fitted a \(\Gamma\) law into the SNP frequency distribution and we kept scaffolds with a SNP frequency under the \(99^{th}\) quantile (\(q_{99} = 0.07810194\)). We thus removed:
- 358 308 SNPs
- including 20 521 transcripts
- representing 1 490 candidate genes
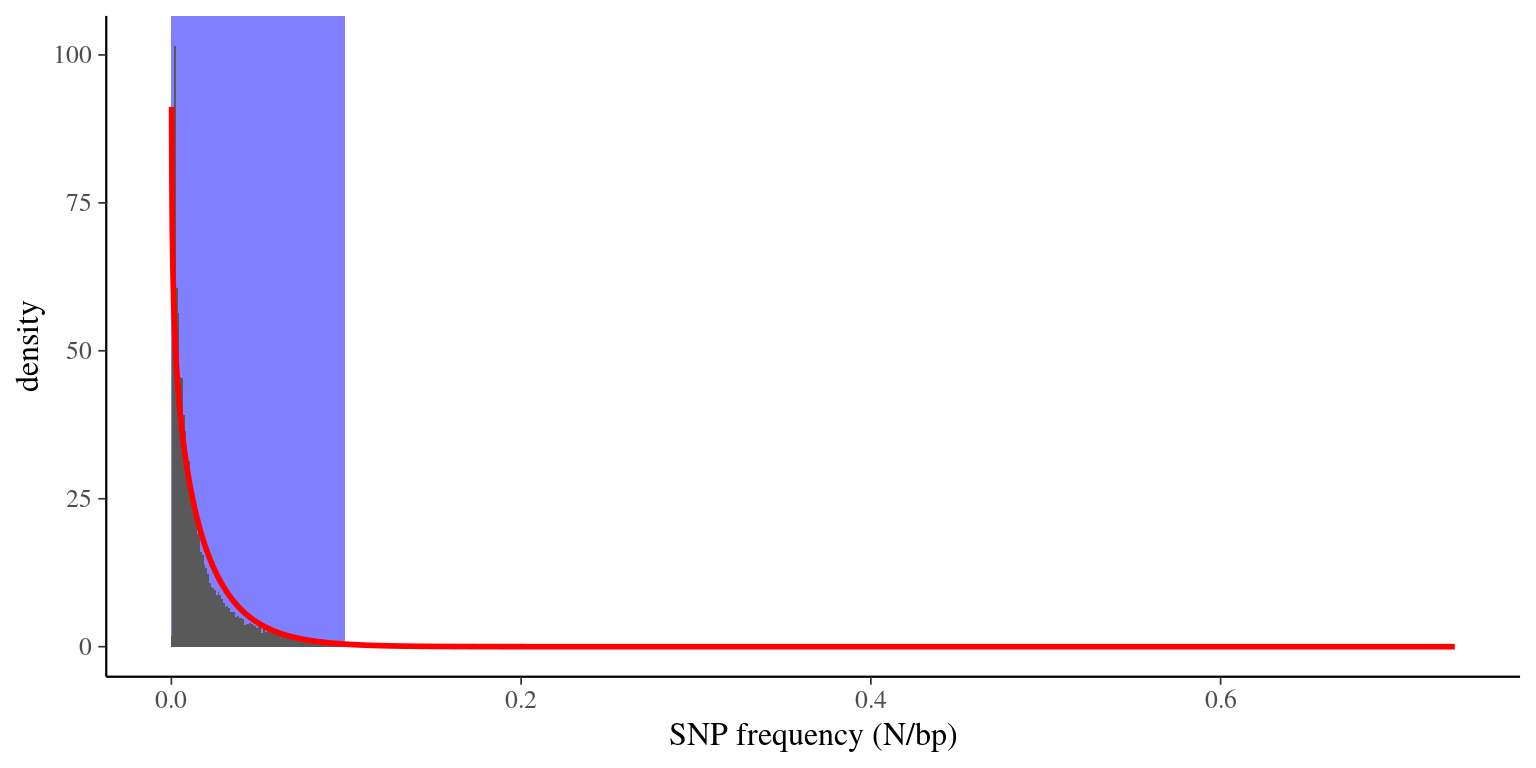
Figure 3.1: Distribution of SNP frequencies in scaffolds. Histogram (gray bars) represents the data, red line represents the Gamma law fit, and blue area represents X*sigma were scaffolds are not excluded.
filtered_data <- snp_genes %>%
filter(freq <= q99) %>%
left_join(data, by = "gene_id") %>%
select(transcript_id, sequence) %>%
unique() %>%
mutate(transcript_id = paste0(">", transcript_id))
filtered_data_fasta <- do.call(rbind, lapply(seq(nrow(filtered_data)),
function(i) t(filtered_data[i, ])))
write.table(filtered_data_fasta, row.names = F, col.names = F, quote = F,
file = file.path(path, "..", "filtered_transcripts.fasta"))3.4 Total filtered transcript
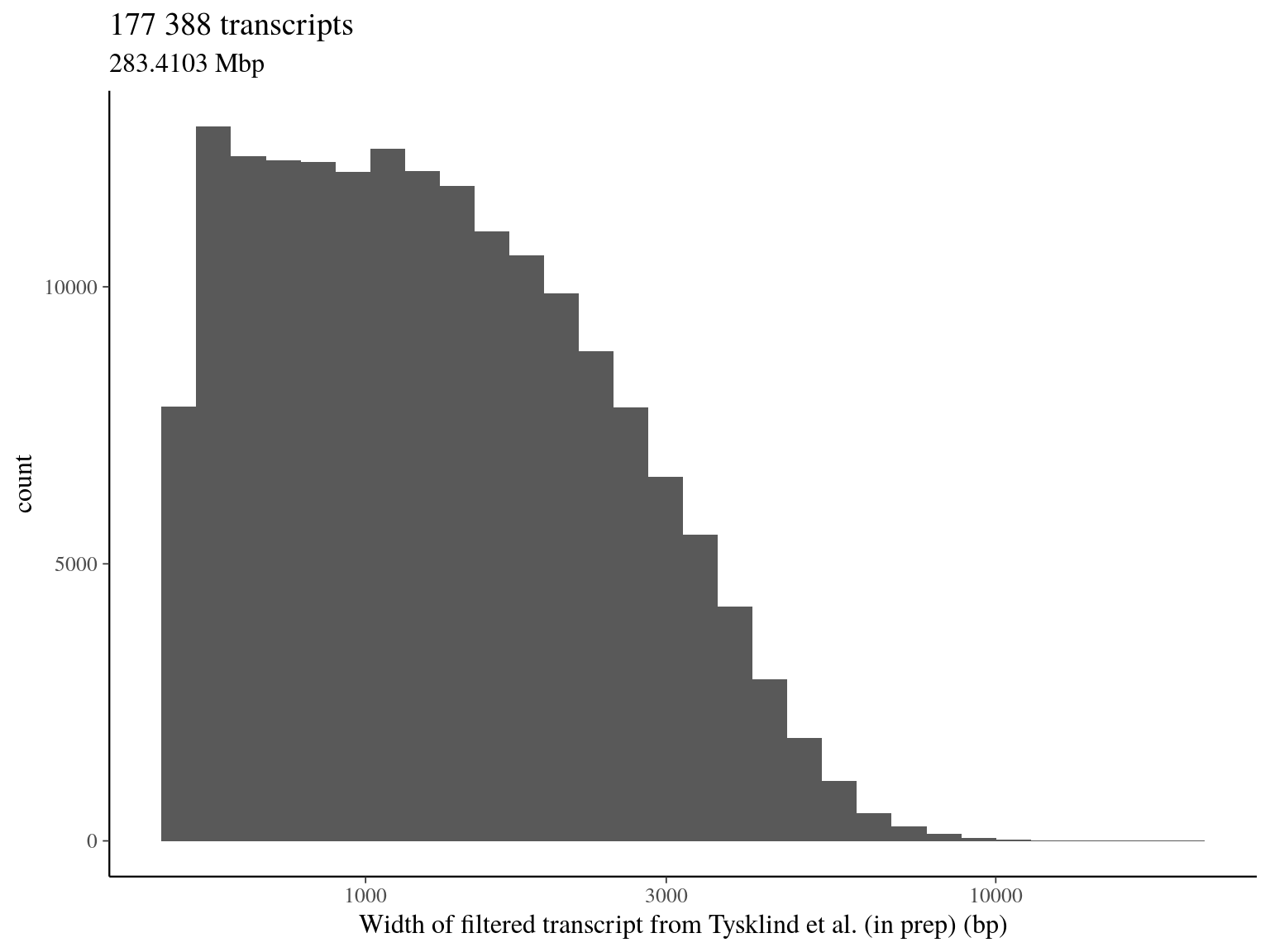
We have a total of:
- 1 382 525 filtered SNPs (over 2 398 550)
- including 177 388 transcripts (over 257 140, including pseudo-genes isoforms)
- representing 63 707 candidate genes (over 76 032)
- for a total of Mbp
References
Lopez-Maestre, H., Brinza, L., Marchet, C., Kielbassa, J., Bastien, S., Boutigny, M., Monnin, D., Filali, A.E., Carareto, C.M., Vieira, C., Picard, F., Kremer, N., Vavre, F., Sagot, M.F. & Lacroix, V. (2016). SNP calling from RNA-seq data without a reference genome: Identification, quantification, differential analysis and impact on the protein sequence. Nucleic Acids Research, 44, gkw655. Retrieved from https://academic.oup.com/nar/article-lookup/doi/10.1093/nar/gkw655