Chapter 4 Neutral region selection
The Guianan sequences from Torroba-Balmor et al (unpublished)…
4.1 Raw reads from Torroba-Balmori et al. (unpublished) alignment on scaffolds from Scotti et al. (in prep)
We will use the French Guianan raw reads from Torroba-Balmori et al. (unpublished), by aligning them on scaffolds from Olsson et al. (2017) with bwa.
#!/bin/bash
#SBATCH --time=36:00:00
#SBATCH -J alignIvan
#SBATCH -o alignIvan_output.out
#SBATCH -e alignIvan_error.out
#SBATCH --mem=20G
#SBATCH --cpus-per-task=1
#SBATCH --mail-type=BEGIN,END,FAIL
# Environment
module purge
module load bioinfo/bwa-0.7.15
module load bioinfo/picard-2.14.1
module load bioinfo/samtools-1.4
module load bioinfo/bedtools-2.26.0
# read preparation
cd ~/work/Symphonia_Torroba/
tar -xvzf Gbs.tar.gz
cd raw
rm PR_49.fastq RG_1.fastq
for file in ./*.fastq
do
echo $file
filename=$(basename "$file")
filename="${filename%.*}"
perl -pe 's|[\h]||g' $file > "${filename}".renamed.fastq
rm $file
done
# variables
cd ~/work/Symphonia_Genomes/Ivan_2018/torroba_alignment
reference=~/work/Symphonia_Genomes/Ivan_2018/merged_1000/merged_1000.fa
query_path=~/work/Symphonia_Torroba/raw
# alignment
bwa index $reference
mkdir bwa
for file in $query_path/*.fastq
do
filename=$(basename "$file")
filename="${filename%.*}"
rg="@RG\tID:${filename}\tSM:${filename}\tPL:IONTORRENT"
bwa mem -M -R "${rg}" $reference $file > bwa/"${filename}.sam"
# rm $file
done
# sam2bam
for file in ./bwa/*.sam
do
filename=$(basename "$file")
filename="${filename%.*}"
java -Xmx4g -jar $PICARD SortSam I=$file O=bwa/"${filename}".bam SORT_ORDER=coordinate
done
# Bam index
for file in bwa/*.bam
do
filename=$(basename "$file")
filename="${filename%.*}"
java -Xmx4g -jar $PICARD BuildBamIndex I=$file O=bwa/"${filename}".bai
done
# sam2bed
mkdir bed
for file in ./bwa/*.bam
do
filename=$(basename "$file")
filename="${filename%.*}"
bedtools bamtobed -i bwa/"${filename}".bam > bed/"${filename}".bed
done
# merge bed
mkdir merged_bed
for file in ./bed/*.bed
do
filename=$(basename "$file")
filename="${filename%.*}"
bedtools merge -i bed/"${filename}".bed > merged_bed/"${filename}".bed
done
cat bed/* | sort -k 1,1 -k2,2n > all.nonunique.bed
bedtools merge -i all.nonunique.bed -c 1 -o count > all.merged.bedbed <- read_tsv(file.path(path, "Ivan_2018", "torroba_alignment", "all.merged.bed"), col_names = F)
names(bed) <- c("scaffold", "start", "end", "coverage")
write_file(paste(unique(bed$scaffold), collapse = "\n"),
file.path(path, "Ivan_2018", "torroba_alignment", "scaffolds.list"))cd ~/Documents/BIOGECO/PhD/data/Symphonia_Genomes/Ivan_2018/torroba_alignment
ref=~/Documents/BIOGECO/PhD/data/Symphonia_Genomes/Ivan_2018/merged_1000/merged_1000.fa
seqtk subseq $ref scaffolds.list >> scaffolds.fa| N | Width (Mbp) | Coverage (%) | |
|---|---|---|---|
| aligned sequence | 1 786 852 | 130.3105 | 29.87923 |
| selected scaffold | 179 665 | 421.7514 | 96.70448 |
| total | 190 098 | 436.1239 | 100.00000 |
4.2 Masking scaffolds with multimatch from Scotti et al. (in prep)
bed <- data.table::fread(file.path(path, "Ivan_2018", "torroba_alignment",
"all.nonunique.bed"), header = F)
names(bed) <- c("scaffold", "start", "end", "read", "quality", "orientation")
multimatch_reads <- bed %>%
filter(duplicated(read)) %>%
select(read) %>%
unique() %>%
unlist()
bed %>%
filter(read %in% multimatch_reads) %>%
write_tsv(file.path(path, "Ivan_2018", "torroba_alignment",
"multimatch.bed"), col_names = F)
rm(bed, multimatch_reads) ; invisible(gc())cd ~/Documents/BIOGECO/PhD/data/Symphonia_Genomes/Ivan_2018/torroba_alignment
bedtools maskfasta -fi scaffolds.fa -bed multimatch.bed -fo masked.scaffolds.fascf <- readDNAStringSet(file.path(path, "Ivan_2018",
"torroba_alignment", "masked.scaffolds.fa"))
scf <- data.frame(scaffold = names(scf), width = width(scf),
N = letterFrequency(scf, letters = "N")) %>%
mutate(Nperc = N/width*100) %>%
filter(Nperc < 25 & width > 1000) %>%
select(scaffold)
write_file(paste(scf$scaffold, collapse = "\n"),
file.path(path, "Ivan_2018", "torroba_alignment", "final.scaffolds.list"))cd ~/Documents/BIOGECO/PhD/data/Symphonia_Genomes/Ivan_2018/torroba_alignment
seqtk subseq masked.scaffolds.fa final.scaffolds.list >> final.scaffolds.fa| N | Width (Mbp) | Mask (%N) | Coverage (%) | |
|---|---|---|---|---|
| selected scaffold | 315 226 | 776.5915 | 6.651114 | 178.0667 |
| total | 190 098 | 436.1239 | NA | 100.0000 |
4.3 Raw reads from Torroba-Balmori et al. (unpublished) alignment on scaffolds from Olsson et al. (2017)
We will again use the French Guianan raw reads from Torroba-Balmori et al. (unpublished), by aligning them on scaffolds from Olsson et al. (2017) with bwa.
#!/bin/bash
#SBATCH --time=36:00:00
#SBATCH -J alignOlsson
#SBATCH -o alignOlsson_output.out
#SBATCH -e alignOlsson_error.out
#SBATCH --mem=20G
#SBATCH --cpus-per-task=1
#SBATCH --mail-type=BEGIN,END,FAIL
# Environment
module purge
module load bioinfo/bwa-0.7.15
module load bioinfo/picard-2.14.1
module load bioinfo/samtools-1.4
module load bioinfo/bedtools-2.26.0
# read preparation
cd ~/work/Symphonia_Torroba/
tar -xvzf Gbs.tar.gz
cd raw
rm PR_49.fastq RG_1.fastq
for file in ./*.fastq
do
echo $file
filename=$(basename "$file")
filename="${filename%.*}"
perl -pe 's|[\h]||g' $file > "${filename}".renamed.fastq
rm $file
done
# variables
cd ~/work/Symphonia_Genomes/Olsson_2016/torroba_alignment
reference=~/work/Symphonia_Genomes/Olsson_2016/Olsson2017/Olsson2017.fa
query_path=~/work/Symphonia_Torroba/raw
# alignment
bwa index $reference
mkdir bwa
for file in $query_path/*.fastq
do
filename=$(basename "$file")
filename="${filename%.*}"
rg="@RG\tID:${filename}\tSM:${filename}\tPL:IONTORRENT"
bwa mem -M -R "${rg}" $reference $file > bwa/"${filename}.sam"
rm $file
done
# sam2bam
for file in ./bwa/*.sam
do
filename=$(basename "$file")
filename="${filename%.*}"
java -Xmx4g -jar $PICARD SortSam I=$file O=bwa/"${filename}".bam SORT_ORDER=coordinate
done
# Bam index
for file in bwa/*.bam
do
filename=$(basename "$file")
filename="${filename%.*}"
java -Xmx4g -jar $PICARD BuildBamIndex I=$file O=bwa/"${filename}".bai
done
# sam2bed
mkdir bed
for file in ./bwa/*.bam
do
filename=$(basename "$file")
filename="${filename%.*}"
bedtools bamtobed -i bwa/"${filename}".bam > bed/"${filename}".bed
done
# merge bed
mkdir merged_bed
for file in ./bed/*.bed
do
filename=$(basename "$file")
filename="${filename%.*}"
bedtools merge -i bed/"${filename}".bed > merged_bed/"${filename}".bed
done
cat bed/* | sort -k 1,1 -k2,2n > all.nonunique.bed
bedtools merge -i all.nonunique.bed -c 1 -o count > all.merged.bedbed <- read_tsv(file.path(path, "Olsson_2016", "torroba_alignment", "all.merged.bed"), col_names = F)
names(bed) <- c("scaffold", "start", "end", "coverage")
write_file(paste(unique(bed$scaffold), collapse = "\n"),
file.path(path, "Olsson_2016", "torroba_alignment", "scaffolds.list"))cd ~/Documents/BIOGECO/PhD/data/Symphonia_Genomes/Olsson_2016/torroba_alignment
ref=~/Documents/BIOGECO/PhD/data/Symphonia_Genomes/Olsson_2016/Olsson2017/Olsson2017.fa
seqtk subseq $ref scaffolds.list >> scaffolds.fa| N | Width (Mbp) | Coverage (%) | |
|---|---|---|---|
| aligned sequence | 1 786 852 | 130.3105 | 12.68385 |
| selected scaffold | 1 056 548 | 590.1957 | 57.44708 |
| total | 2 653 526 | 1 027.3729 | 100.00000 |
4.4 Masking scaffolds with multimatch from Olsson et al. (2017)
bed <- data.table::fread(file.path(path, "Olsson_2016", "torroba_alignment",
"all.nonunique.bed"), header = F)
names(bed) <- c("scaffold", "start", "end", "read", "quality", "orientation")
multimatch_reads <- bed %>%
filter(duplicated(read)) %>%
select(read) %>%
unique() %>%
unlist()
bed %>%
filter(read %in% multimatch_reads) %>%
write_tsv(file.path(path, "Olsson_2016", "torroba_alignment",
"multimatch.bed"), col_names = F)
rm(bed, multimatch_reads) ; invisible(gc())cd ~/Documents/BIOGECO/PhD/data/Symphonia_Genomes/Olsson_2016/torroba_alignment
bedtools maskfasta -fi scaffolds.fa -bed multimatch.bed -fo masked.scaffolds.fascf <- readDNAStringSet(file.path(path, "Olsson_2016",
"torroba_alignment", "masked.scaffolds.fa"))
scf <- data.frame(scaffold = names(scf), width = width(scf),
N = letterFrequency(scf, letters = "N")) %>%
mutate(Nperc = N/width*100) %>%
filter(Nperc < 25 & width > 1000) %>%
select(scaffold)
write_file(paste(scf$scaffold, collapse = "\n"),
file.path(path, "Olsson_2016", "torroba_alignment", "final.scaffolds.list"))cd ~/Documents/BIOGECO/PhD/data/Symphonia_Genomes/Olsson_2016/torroba_alignment
ref=~/Documents/BIOGECO/PhD/data/Symphonia_Genomes/Olsson_2016/Olsson2017/Olsson2017.fa
seqtk subseq masked.scaffolds.fa final.scaffolds.list >> final.scaffolds.fa| N | Width (Mbp) | Mask (%N) | Coverage (%) | |
|---|---|---|---|---|
| selected scaffold | 245 646 | 464.1304 | 1.896894 | 45.17643 |
| total | 2 653 526 | 1 027.3729 | NA | 100.00000 |
4.5 Removing scaffolds already matching transcripts
func <-unlist(read_tsv(file.path(path, "Ivan_2018",
"transcript_alignment", "selected_scaffolds.list"), col_names = F))
neutral <- readDNAStringSet(file.path(path, "Ivan_2018", "torroba_alignment",
"final.scaffolds.fa"))
writeXStringSet(neutral[setdiff(names(neutral), func)], file.path(path, "neutral_selection", "Ivan.selected.scaffolds.fa"))func <-unlist(read_tsv(file.path(path, "Olsson_2016",
"transcript_alignment", "selected_scaffolds.list"), col_names = F))
neutral <- readDNAStringSet(file.path(path, "Olsson_2016", "torroba_alignment",
"final.scaffolds.fa"))
writeXStringSet(neutral[setdiff(names(neutral), func)], file.path(path, "neutral_selection", "Olsson.selected.scaffolds.fa"))4.6 Merge of selected scaffolds
We merged selected scaffolds from Scotti et al (in prep) and Olsson et al. (2017) with quickmerge.
cd ~/Documents/BIOGECO/PhD/data/Symphonia_Genomes/neutral_selection
ref=Ivan.selected.scaffolds.fa
query=Olsson.selected.scaffolds.fa
nucmer -l 100 -prefix out $ref $query
delta-filter -i 95 -r -q out.delta > out.rq.delta
~/Tools/quickmerge/quickmerge -d out.rq.delta -q $query -r $ref -hco 5.0 -c 1.5 -l n -ml mWe merged selected scaffolds from Scotti et al (in prep) and Olsson et al. (2017) with quickmerge. We found 13 343 overlaps resulting a final merged assembly of 82 792 scaffolds for a total lenght of 146.80 Mb.
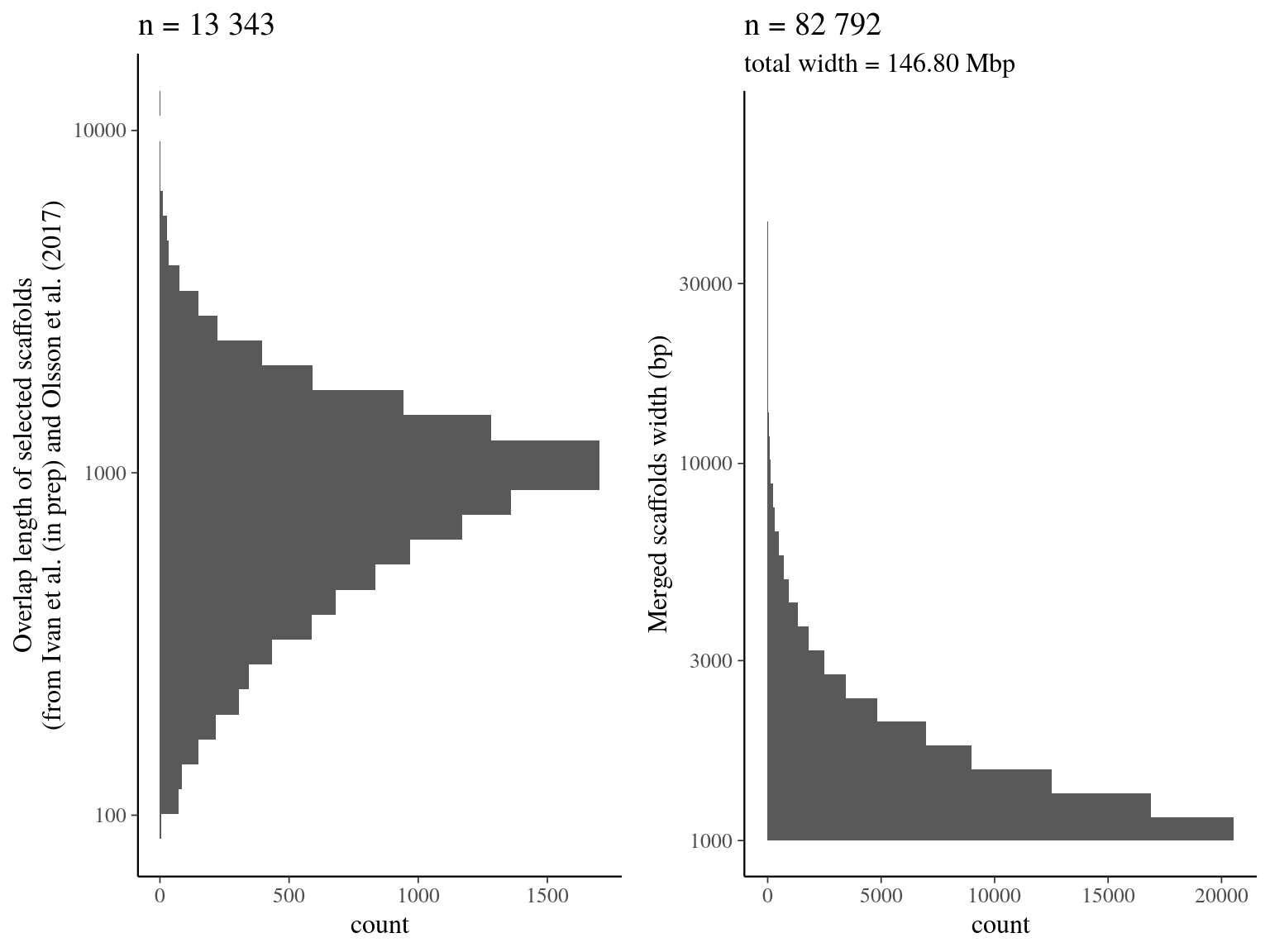
Figure 4.1: Merging result from quickmerge. Left graph represents the overlap distribution. Right graph represent the merged scaffolds distribution.
4.7 Final subset of selected neutral scaffolds
We finally selected 0.533 Mb of sequences by sampling 533 1-kb sequences among 533 scaffolds (1 sequence per scaffold) with a probability \(p=\frac{scaffold\_length}{total\_length}\).
scf <- readDNAStringSet(file.path(path, "neutral_selection", "merged.fasta"))
selection <- data.frame(scf = names(scf), width = width(scf),
N = letterFrequency(scf, "N")) %>%
sample_n(533, weight = width) %>%
select(scf) %>%
unlist()
scf_sel <- subseq(scf[selection], end=1000, width=1000)
writeXStringSet(scf_sel, file.path(path, "neutral_selection", "selected.scaffolds.fa"))| N | Width (Mbp) | Mask (%N) |
|---|---|---|
| 533 | 0.533 | 0.0042383 |
4.8 Repetitive regions final check
Last but not least, we do not want to include repetitive regions in our targets for baits design. We consequently aligned raw reads from one library from Scotti et al. (in prep) on our targets with bwa.
cd ~/Documents/BIOGECO/PhD/data/Symphonia_Genomes/neutral_selection
reference=selected.scaffolds.fa
query=~/Documents/BIOGECO/PhD/data/Symphonia_Genomes/Ivan_2018/raw_reads/globu1_symphonia_globulifera_CTTGTA_L001_R1_001.fastq.gz
bwa index $reference
bwa mem -M $reference $query > raw_read_alignment.sam
picard=~/Tools/picard/picard.jar
java -Xmx4g -jar $picard SortSam I=raw_read_alignment.sam O=raw_read_alignment.bam SORT_ORDER=coordinate
bedtools bamtobed -i raw_read_alignment.bam > raw_read_alignment.bed
cat raw_read_alignment.bed | sort -k 1,1 -k2,2n > raw_read_alignment.sorted.bed
bedtools merge -i raw_read_alignment.sorted.bed -c 1 -o count > raw_read_alignment.merged.bedWe obtained a continuous decreasing distribution of read coverage across our scaffolds regions (figure 4.2). We fitted a \(\Gamma\) distribution with positive parameters for scaffolds regions with a coverage under 5 000 (non continuous distribution with optimization issues). We obtained a distribution with a mean of 324 reads per region and a \(99^{th}\) quantile of 4 042. We decided to mask regions with a coverage over the \(99^{th}\) quantile and remove scaffolds with a mask superior to 75% of its total length (figure ??).
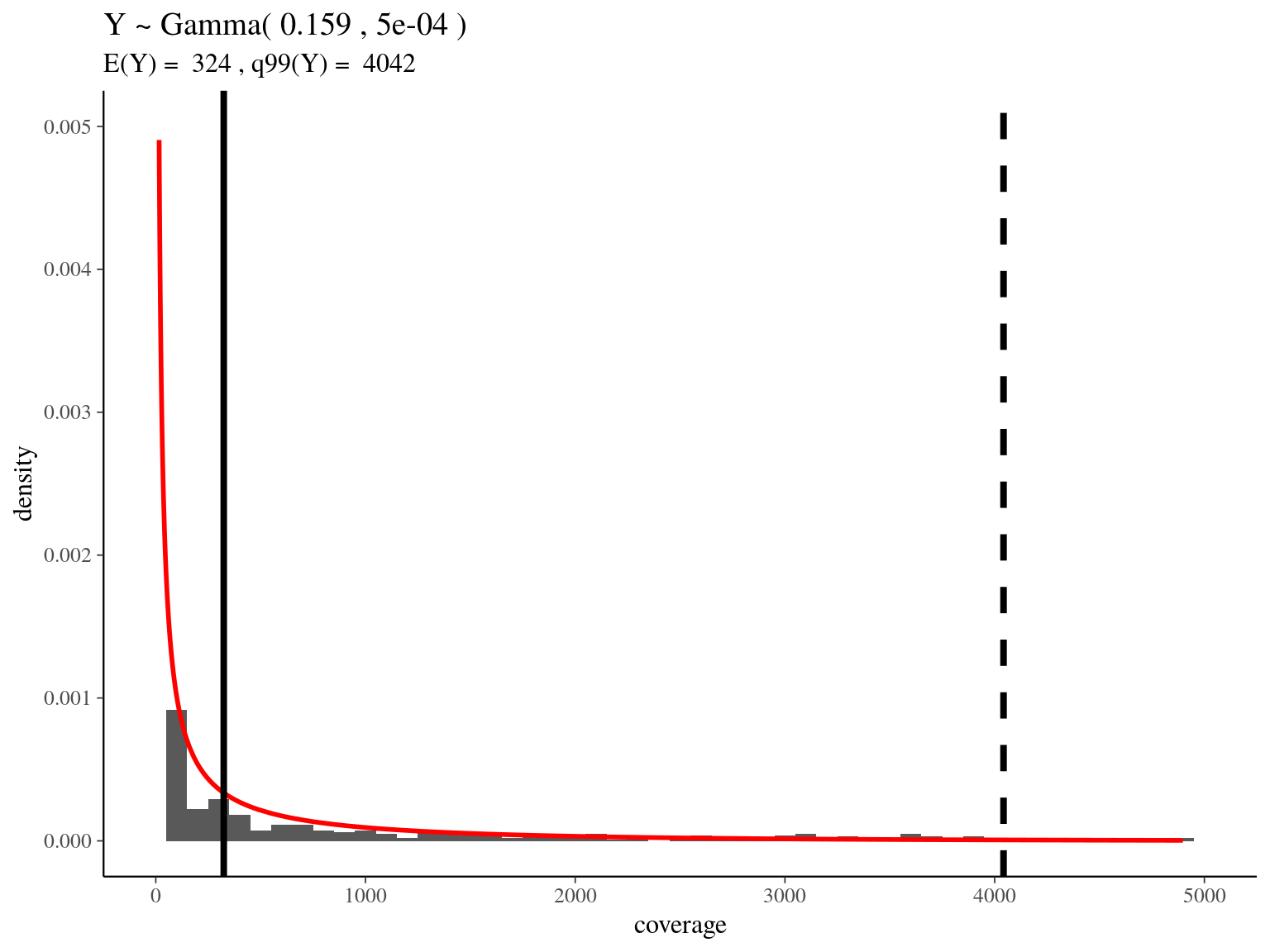
Figure 4.2: Read coverage distribution.
repetitive_target <- bed %>%
filter(coverage > qgamma(0.99, alpha, beta)) %>%
mutate(size = end - start)
repetitive_target %>%
select(target, start, end) %>%
arrange(target, start, end) %>%
mutate_if(is.numeric, as.character) %>%
write_tsv(path = file.path(path, "neutral_selection", "repetitive_targets.bed"),
col_names = F)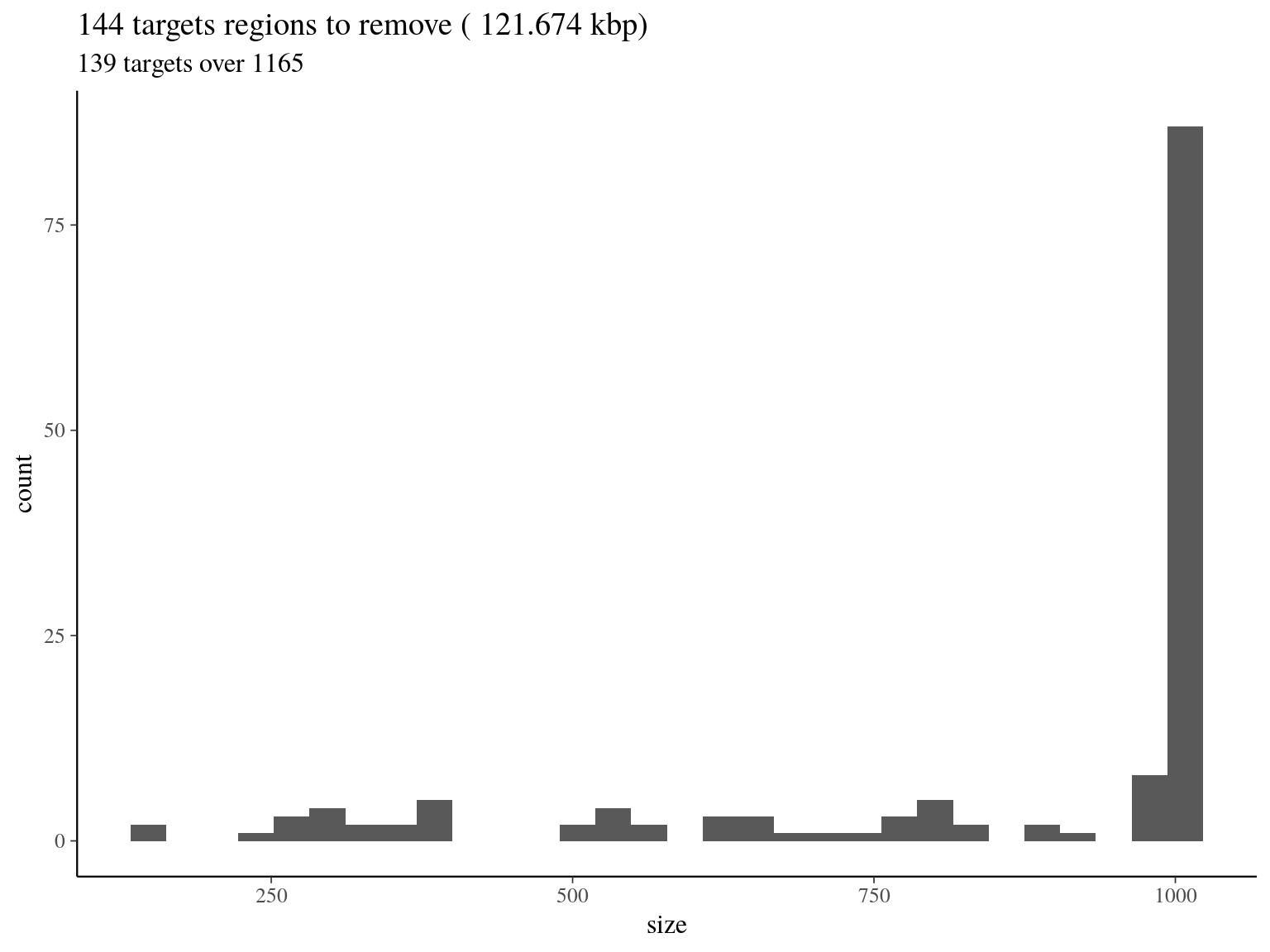
Figure 4.3: target regions with a coverage over the 99th quantile of the fitted Gamma distribution (4042).
cd ~/Documents/BIOGECO/PhD/data/Symphonia_Genomes/neutral_selection
cat repetitive_targets.bed | sort -k 1,1 -k2,2n > repetitive_targets.sorted.bed
bedtools maskfasta -fi selected.scaffolds.fa -bed repetitive_targets.sorted.bed -fo targets.masked.fastatargets <- readDNAStringSet(file.path(path, "neutral_selection", "targets.masked.fasta"))
writeXStringSet(targets[which(letterFrequency(targets, "N")/width(targets) < 0.65)],
file.path(path, "neutral_selection", "targets.filtered.masked.fasta"))| N | Width (Mbp) | Mask (%N) |
|---|---|---|
| 415 | 0.415 | 0.024412 |
References
Olsson, S., Seoane-Zonjic, P., Bautista, R., Claros, M.G., González-Martínez, S.C., Scotti, I., Scotti-Saintagne, C., Hardy, O.J. & Heuertz, M. (2017). Development of genomic tools in a widespread tropical tree, Symphonia globulifera L.f.: a new low-coverage draft genome, SNP and SSR markers. Molecular Ecology Resources, 17, 614–630. Retrieved from http://doi.wiley.com/10.1111/1755-0998.12605